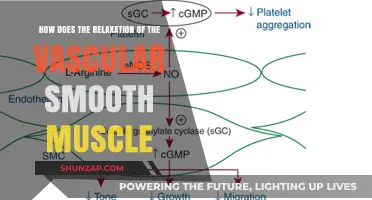
Nitric oxide (NO) is a crucial signaling molecule in the human body that plays a significant role in regulating vascular tone and blood flow by inducing smooth muscle relaxation. When produced by endothelial cells lining blood vessels, NO diffuses into adjacent smooth muscle cells, where it activates the enzyme soluble guanylate cyclase (sGC). This activation leads to an increase in cyclic guanosine monophosphate (cGMP) levels, which in turn triggers a cascade of events, including the dephosphorylation of myosin light chains by protein phosphatase 1. This process reduces the interaction between actin and myosin filaments, causing the smooth muscle cells to relax and the blood vessels to dilate, thereby improving blood flow and reducing blood pressure. This mechanism underscores NO’s vital role in cardiovascular health and its therapeutic potential in treating conditions like hypertension and erectile dysfunction.
| Characteristics | Values |
|---|---|
| Mechanism of Action | Nitric oxide (NO) activates soluble guanylate cyclase (sGC) in smooth muscle cells, leading to increased cyclic guanosine monophosphate (cGMP) production. |
| cGMP Role | cGMP activates protein kinase G (PKG), which phosphorylates target proteins, reducing intracellular calcium levels and promoting smooth muscle relaxation. |
| Calcium Regulation | NO-induced PKG activation decreases calcium influx via voltage-gated calcium channels and enhances calcium reuptake by the sarcoplasmic reticulum, lowering cytosolic calcium. |
| Myofilament Interaction | Reduced calcium levels decrease calcium binding to calmodulin, inhibiting myosin light chain kinase (MLCK) and reducing myosin light chain phosphorylation, leading to muscle relaxation. |
| Vasodilation | In vascular smooth muscle, NO-mediated relaxation causes vasodilation, increasing blood flow and reducing blood pressure. |
| cGMP Degradation | Phosphodiesterase (PDE) enzymes degrade cGMP, terminating the relaxation signal. PDE5 inhibitors (e.g., sildenafil) enhance NO-cGMP signaling by inhibiting cGMP breakdown. |
| Clinical Relevance | NO pathway dysfunction is implicated in hypertension, erectile dysfunction, and other vascular disorders. Therapies targeting this pathway (e.g., nitrates, PDE5 inhibitors) are used to treat these conditions. |
| Endothelial Dependency | Endothelial cells produce NO via endothelial nitric oxide synthase (eNOS), which diffuses to adjacent smooth muscle cells to initiate relaxation. |
| Second Messenger | cGMP acts as a second messenger in NO signaling, transducing the extracellular NO signal into intracellular responses. |
| Reversibility | The relaxation effect is reversible, as cGMP levels return to baseline upon cessation of NO production or increased PDE activity. |
Explore related products






What You'll Learn
![]()
cGMP signaling pathway activation
Nitric oxide (NO) is a potent vasodilator that relaxes smooth muscle cells in blood vessels, primarily through the activation of the cGMP signaling pathway. This process begins when NO diffuses into the smooth muscle cells and binds to the heme moiety of soluble guanylate cyclase (sGC), an enzyme that catalyzes the conversion of guanosine triphosphate (GTP) to cyclic guanosine monophosphate (cGMP). The binding of NO to sGC induces a conformational change, activating the enzyme and increasing cGMP production. This second messenger, cGMP, then triggers a cascade of events leading to smooth muscle relaxation.
Mechanism Unveiled: A Step-by-Step Breakdown
- NO Production and Release: Endothelial cells lining blood vessels produce NO via the enzyme endothelial nitric oxide synthase (eNOS). Stimuli such as shear stress or acetylcholine trigger eNOS activation, releasing NO into the surrounding tissue.
- SGC Activation: NO binds to sGC, causing it to shed its inhibitory subunit and become fully active. This activation is rapid, occurring within seconds of NO binding.
- CGMP Synthesis: Activated sGC catalyzes the conversion of GTP to cGMP, elevating intracellular cGMP levels.
- Protein Kinase G (PKG) Activation: cGMP binds to PKG, a cGMP-dependent protein kinase, causing its activation. PKG phosphorylates target proteins, initiating the relaxation process.
- Smooth Muscle Relaxation: PKG-mediated phosphorylation leads to decreased calcium sensitivity in the contractile machinery of smooth muscle cells. Specifically, it reduces calcium release from the sarcoplasmic reticulum and inhibits calcium binding to calmodulin, resulting in decreased myosin light chain phosphorylation and muscle relaxation.
Clinical Relevance and Practical Applications
Understanding the cGMP signaling pathway has led to the development of therapeutic agents like phosphodiesterase type 5 (PDE5) inhibitors (e.g., sildenafil, tadalafil). These drugs block the breakdown of cGMP, prolonging its effects and enhancing vasodilation. For instance, in patients with erectile dysfunction or pulmonary hypertension, PDE5 inhibitors are prescribed at dosages ranging from 20 to 60 mg, depending on age, comorbidities, and response. Clinicians must monitor for side effects such as hypotension, particularly in older adults or those on concurrent antihypertensive medications.
Comparative Insights: cGMP vs. Other Pathways
While cGMP signaling is central to NO-mediated smooth muscle relaxation, other pathways, such as the β-adrenergic pathway, also induce relaxation but via cAMP. The cGMP pathway is uniquely sensitive to NO and sGC modulators, making it a targeted therapeutic avenue. Unlike cAMP, which primarily activates protein kinase A (PKA), cGMP’s activation of PKG offers a distinct regulatory mechanism, highlighting the specificity of NO’s action in vascular physiology.
Optimizing cGMP Signaling: Tips and Cautions
To enhance cGMP signaling naturally, lifestyle modifications such as regular exercise, a nitrate-rich diet (e.g., beets, spinach), and stress reduction can boost NO production. However, excessive supplementation with NO precursors (e.g., L-arginine) may be counterproductive, as high doses can lead to oxidative stress. For patients on PDE5 inhibitors, grapefruit consumption should be avoided, as it inhibits drug metabolism, increasing the risk of adverse effects. Always consult a healthcare provider before initiating new treatments or supplements.
Alcohol's Effect on Facial Muscles: Does It Truly Induce Relaxation?
You may want to see also
Explore related products






![]()
Inhibition of calcium influx in cells
Nitric oxide (NO) is a potent vasodilator that plays a critical role in relaxing smooth muscle cells, particularly in blood vessels. One of its primary mechanisms of action involves the inhibition of calcium influx into these cells, a process essential for understanding its therapeutic potential. Calcium ions (Ca²⁺) are key mediators of smooth muscle contraction, and their entry into cells is tightly regulated by voltage-gated calcium channels (VGCCs). When NO is produced—often in response to endothelial stimulation—it diffuses into adjacent smooth muscle cells and activates soluble guanylate cyclase (sGC), leading to increased cyclic guanosine monophosphate (cGMP) levels. This second messenger then activates protein kinase G (PKG), which phosphorylates and inhibits VGCCs, thereby reducing calcium influx.
Consider the step-by-step process: NO production triggers a cascade that ultimately suppresses calcium entry, preventing the activation of calmodulin and myosin light chain kinase (MLCK), enzymes critical for muscle contraction. Without sufficient calcium, the contractile machinery remains inactive, leading to relaxation. This mechanism is particularly relevant in conditions like hypertension, where excessive smooth muscle tone restricts blood flow. For instance, nitroglycerin, a common angina medication, works by releasing NO, which dilates coronary arteries via this calcium-inhibiting pathway. Dosage typically ranges from 0.3 to 0.6 mg sublingually, with effects lasting 30–60 minutes, highlighting the rapid and targeted nature of NO’s action.
From a comparative perspective, the inhibition of calcium influx by NO contrasts with other vasodilators like calcium channel blockers (e.g., nifedipine), which directly block VGCCs. While both approaches reduce calcium levels, NO’s indirect method via cGMP/PKG signaling offers additional benefits, such as improved endothelial function and reduced oxidative stress. However, NO’s short half-life (seconds) and susceptibility to degradation by reactive oxygen species (ROS) necessitate careful administration, especially in older adults or those with cardiovascular risk factors. Practical tips include avoiding high-fat meals, which can impair NO bioavailability, and monitoring for hypotensive effects, particularly in patients with autonomic dysfunction.
Analytically, the inhibition of calcium influx by NO underscores its role as a physiological regulator of vascular tone. Studies in animal models have shown that NO deficiency, often seen in conditions like atherosclerosis, leads to heightened calcium sensitivity and vasoconstriction. Conversely, therapies aimed at enhancing NO bioavailability, such as dietary nitrate supplementation (e.g., beetroot juice, 70–140 mmol/day), have demonstrated improvements in endothelial function and reduced blood pressure in adults over 50. This evidence supports the idea that modulating calcium influx via NO pathways is a viable strategy for managing vascular disorders.
In conclusion, the inhibition of calcium influx by NO is a precise and effective mechanism for smooth muscle relaxation, with broad implications for cardiovascular health. By understanding this process, clinicians and researchers can better leverage NO-based therapies, from acute treatments like nitroglycerin to lifestyle interventions like nitrate-rich diets. However, individualized approaches are essential, considering factors like age, comorbidities, and medication interactions to maximize benefits while minimizing risks. This targeted modulation of calcium signaling exemplifies NO’s unique role in maintaining vascular homeostasis.
Gabapentin for Muscle Relaxation: How Effective Is It?
You may want to see also
Explore related products






![]()
Protein kinase G (PKG) role
Nitric oxide (NO) is a potent vasodilator, crucial for relaxing smooth muscle in blood vessels, a process central to regulating blood flow and pressure. At the heart of this mechanism lies Protein Kinase G (PKG), a key mediator of NO’s effects. When NO is produced, it diffuses into smooth muscle cells and activates soluble guanylate cyclase (sGC), which converts guanosine triphosphate (GTP) into cyclic guanosine monophosphate (cGMP). This cGMP then binds to PKG, triggering its activation. Once activated, PKG initiates a cascade of phosphorylation events that ultimately lead to smooth muscle relaxation.
To understand PKG’s role, consider its targets within the cell. PKG phosphorylates proteins like myosin phosphatase target subunit 1 (MYPT1), which inhibits the activity of myosin light chain phosphatase. This inhibition reduces the phosphorylation of myosin light chains, weakening the actin-myosin interactions essential for muscle contraction. Additionally, PKG activates potassium channels, increasing potassium efflux and hyperpolarizing the cell membrane. This hyperpolarization reduces calcium influx, lowering intracellular calcium levels and further inhibiting muscle contraction. Together, these actions promote smooth muscle relaxation.
Practical implications of PKG’s role are evident in therapeutic applications. For instance, drugs like nitroglycerin, used to treat angina, work by releasing NO, which activates the PKG pathway. Dosage is critical; nitroglycerin is typically administered sublingually at 0.3–0.6 mg every 5 minutes, up to 3 doses, to avoid hypotension. Similarly, phosphodiesterase type 5 (PDE5) inhibitors, such as sildenafil, enhance PKG activity by preventing cGMP breakdown, making them effective in treating erectile dysfunction and pulmonary hypertension. However, caution is advised in patients with cardiovascular conditions, as excessive PKG activation can lead to severe hypotension.
Comparatively, PKG’s role in smooth muscle relaxation contrasts with pathways involving Protein Kinase A (PKA), which is activated by cyclic adenosine monophosphate (cAMP). While both kinases promote relaxation, PKG is specifically tied to NO signaling, whereas PKA is associated with catecholamines and prostacyclin. This distinction highlights PKG’s unique importance in vascular physiology. Dysregulation of PKG, such as in cases of sGC or PKG mutations, can lead to hypertension or impaired blood flow, underscoring its critical role in maintaining vascular health.
In summary, PKG is a linchpin in NO-mediated smooth muscle relaxation, acting through precise phosphorylation events and ion channel modulation. Its activation is harnessed in therapies for cardiovascular and pulmonary disorders, but careful dosing and patient selection are essential. Understanding PKG’s mechanisms not only elucidates vascular physiology but also informs targeted interventions for conditions where smooth muscle function is compromised.
Does Bentyl Relax the Heart Muscle? Exploring Its Effects and Safety
You may want to see also
Explore related products






![]()
Phosphodiesterase inhibition mechanism
Nitric oxide (NO) is a potent vasodilator that relaxes smooth muscle cells primarily by activating soluble guanylate cyclase (sGC), which increases cyclic guanosine monophosphate (cGMP) levels. However, the efficacy of this relaxation depends on how long cGMP remains active within the cell. This is where phosphodiesterase (PDE) inhibition plays a critical role. PDE enzymes degrade cGMP, limiting its signaling duration and, consequently, the extent of smooth muscle relaxation. By inhibiting PDE, particularly PDE5, the breakdown of cGMP is slowed, prolonging its effects and enhancing NO-mediated vasodilation.
Consider the mechanism in a step-by-step manner. First, NO binds to sGC, stimulating cGMP production. Next, cGMP activates protein kinase G (PKG), which phosphorylates target proteins, leading to smooth muscle relaxation. Without PDE inhibition, PDE5 rapidly hydrolyzes cGMP to inactive GMP, curtailing the relaxation response. Inhibiting PDE5, as seen with drugs like sildenafil (25–100 mg dosage, typically 1 hour before activity), preserves cGMP levels, allowing sustained PKG activation and prolonged smooth muscle relaxation. This mechanism is particularly relevant in treating conditions like erectile dysfunction and pulmonary hypertension.
Analyzing the clinical implications, PDE5 inhibitors are not universally effective. For instance, in patients with severe endothelial dysfunction, NO production may be insufficient to initiate the cGMP pathway, rendering PDE inhibition less effective. Additionally, PDE5 inhibitors can cause side effects such as headaches, flushing, and hypotension, especially in older adults (>65 years) or those on nitrate medications. Thus, while PDE inhibition amplifies NO’s effects, it is a downstream solution that relies on adequate NO bioavailability.
Comparatively, PDE inhibition contrasts with other strategies to enhance NO signaling, such as increasing NO production via L-arginine supplementation or using sGC stimulators. PDE inhibitors act by preserving the existing cGMP signal, whereas sGC stimulators directly enhance cGMP synthesis, even in the presence of dysfunctional NO production. For example, riociguat, an sGC stimulator, is used in pulmonary arterial hypertension, while PDE5 inhibitors like tadalafil (5–20 mg daily) are preferred for erectile dysfunction. The choice depends on the underlying pathology and desired duration of action.
In practical terms, optimizing PDE inhibition requires careful consideration of timing, dosage, and patient profile. For erectile dysfunction, sildenafil should be taken on an empty stomach to maximize absorption, while tadalafil’s longer half-life (17.5 hours) allows for daily dosing. In pulmonary hypertension, combining PDE5 inhibitors with calcium channel blockers or prostacyclin analogs may enhance efficacy but increases the risk of hypotension. Monitoring for drug interactions, particularly with nitrates, is essential to prevent severe hypotensive episodes. By understanding the PDE inhibition mechanism, clinicians can tailor therapies to maximize smooth muscle relaxation while minimizing risks.
Quetiapine and Muscle Relaxation: Understanding Its Effects on the Body
You may want to see also
Explore related products






![]()
Smooth muscle cell hyperpolarization effect
Nitric oxide (NO) is a potent vasodilator that relaxes smooth muscle cells primarily by activating soluble guanylate cyclase (sGC), increasing cyclic guanosine monophosphate (cGMP), and subsequently activating protein kinase G (PKG). However, another critical mechanism involves the hyperpolarization of smooth muscle cells, which further enhances relaxation. This process is mediated by the opening of potassium channels, particularly the large-conductance calcium-activated potassium (BKCa) channels, leading to an efflux of potassium ions and membrane hyperpolarization.
To understand the hyperpolarization effect, consider the electrical state of smooth muscle cells. In a contracted state, the cell membrane is depolarized, allowing calcium influx through voltage-gated calcium channels. This intracellular calcium binds to calmodulin, activating myosin light-chain kinase (MLCK) and promoting muscle contraction. NO disrupts this cycle by triggering a cascade that ultimately opens BKCa channels. These channels are sensitive to both calcium and voltage, and their activation leads to potassium efflux, repolarizing the membrane and reducing calcium influx. This decrease in intracellular calcium concentration inhibits MLCK, leading to smooth muscle relaxation.
Practical implications of this mechanism are evident in pharmacological interventions. For instance, drugs like sildenafil and tadalafil enhance cGMP levels by inhibiting phosphodiesterase type 5 (PDE5), indirectly supporting hyperpolarization by prolonging the effects of NO. Additionally, BKCa channel openers, such as NS1619, are being explored as potential therapies for hypertension and erectile dysfunction. Dosage considerations vary; for example, sildenafil is typically prescribed at 25–100 mg, taken 30–60 minutes before activity, while BKCa activators are still in experimental stages with dosages dependent on specific formulations.
A comparative analysis highlights the synergy between cGMP-mediated and hyperpolarization-mediated pathways. While cGMP activation directly reduces calcium sensitivity through PKG, hyperpolarization acts upstream by limiting calcium entry. This dual action explains why NO is such an effective vasodilator. For instance, in aging populations (over 65), endothelial dysfunction often reduces NO bioavailability, impairing both pathways and contributing to vascular stiffness. Supplementing with NO precursors like L-arginine (3–6 grams daily) or beetroot juice (rich in nitrates) can partially restore these mechanisms, though individual responses vary based on health status and comorbidities.
In conclusion, the hyperpolarization effect of NO on smooth muscle cells is a critical yet often overlooked component of its vasodilatory action. By targeting potassium channels, particularly BKCa, NO ensures robust and sustained relaxation. This mechanism not only complements the cGMP pathway but also offers therapeutic opportunities for conditions characterized by impaired vascular function. Understanding this process allows for more targeted interventions, whether through pharmacological agents or lifestyle modifications, to optimize vascular health across diverse age groups and medical conditions.
Muscle Energy Mechanics: Fueling Contraction and Relaxation Processes
You may want to see also
Frequently asked questions
Nitric oxide activates soluble guanylate cyclase (sGC) in smooth muscle cells, increasing cyclic guanosine monophosphate (cGMP) levels. cGMP then activates protein kinase G (PKG), which phosphorylates target proteins, leading to decreased calcium sensitivity and relaxation of smooth muscle.
NO reduces calcium influx and release from intracellular stores by inhibiting calcium channels and modulating calcium-handling proteins. Lower intracellular calcium levels decrease myosin light chain phosphorylation, resulting in smooth muscle relaxation.
NO-mediated smooth muscle relaxation is crucial in vasodilation (widening blood vessels), bronchial dilation (opening airways), and gastrointestinal motility. It also plays a role in penile erection by relaxing corporal smooth muscle.