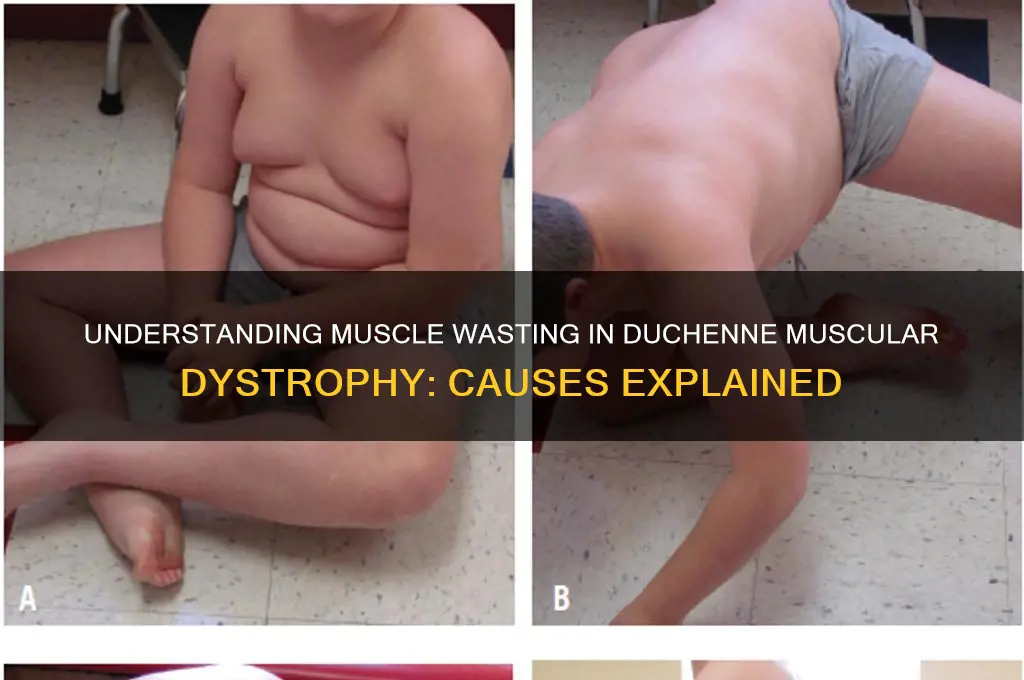
Duchenne Muscular Dystrophy (DMD) is a severe genetic disorder characterized by progressive muscle weakness and wasting due to mutations in the dystrophin gene, which encodes a protein essential for muscle fiber integrity. In individuals with DMD, the absence or dysfunction of dystrophin leads to repeated cycles of muscle damage and repair, ultimately resulting in muscle fiber necrosis, fibrosis, and fatty infiltration. This chronic degeneration is exacerbated by inflammation, oxidative stress, and impaired satellite cell function, which hinder effective muscle regeneration. Additionally, the loss of dystrophin disrupts the dystrophin-glycoprotein complex, compromising the sarcolemma’s stability and increasing susceptibility to mechanical stress during muscle contraction. Over time, these cumulative effects lead to irreversible muscle wasting, loss of ambulation, and systemic complications, making DMD a devastating condition with significant impacts on quality of life.
| Characteristics | Values |
|---|---|
| Underlying Cause | Deficiency of dystrophin protein due to mutations in the DMD gene. |
| Dystrophin Function | Acts as a shock absorber and stabilizes muscle fibers during contraction. |
| Consequence of Dystrophin Deficiency | Increased membrane fragility, leading to repeated muscle fiber damage. |
| Mechanisms of Muscle Wasting | 1. Repeated Cycles of Damage and Repair: Chronic inflammation and fibrosis replace functional muscle tissue. 2. Calcium Dysregulation: Elevated intracellular calcium activates proteases and apoptosis pathways. 3. Oxidative Stress: Accumulation of reactive oxygen species (ROS) damages muscle cells. 4. Impaired Regeneration: Reduced satellite cell function limits muscle repair. 5. Fibrosis: Excessive extracellular matrix deposition replaces muscle fibers. |
| Secondary Factors | Chronic inflammation, mitochondrial dysfunction, and metabolic abnormalities contribute to progressive wasting. |
| Clinical Progression | Muscle wasting begins in early childhood, progressively affecting proximal muscles first, leading to loss of ambulation and respiratory/cardiac involvement. |
Explore related products






What You'll Learn
- Genetic mutations in dystrophin gene disrupt muscle fiber function, leading to progressive weakness and wasting
- Repeated muscle damage and repair cycles cause fibrosis, replacing functional muscle with non-contractile tissue
- Chronic inflammation accelerates muscle breakdown and impairs regeneration in DMD patients over time
- Lack of dystrophin protein increases muscle membrane fragility, causing frequent injury and degeneration
- Oxidative stress and mitochondrial dysfunction contribute to muscle cell death and atrophy in DMD
![]()
Genetic mutations in dystrophin gene disrupt muscle fiber function, leading to progressive weakness and wasting
Duchenne muscular dystrophy (DMD) is a severe genetic disorder primarily characterized by progressive muscle weakness and wasting. At the core of this condition are genetic mutations in the dystrophin gene, which plays a critical role in maintaining the structural integrity of muscle fibers. The dystrophin gene is responsible for producing dystrophin, a protein that acts as a vital link between the internal cytoskeleton of muscle cells and the extracellular matrix. This linkage is essential for stabilizing muscle fibers during contraction and preventing damage. When mutations occur in the dystrophin gene, they often result in the production of a nonfunctional or absent dystrophin protein, leading to the hallmark symptoms of DMD.
The absence or dysfunction of dystrophin disrupts the normal mechanics of muscle fibers, making them highly susceptible to mechanical stress during movement. In healthy muscles, dystrophin absorbs the force generated during muscle contraction, protecting the sarcolemma (the cell membrane of muscle fibers) from tearing. However, in individuals with DMD, the lack of functional dystrophin causes the sarcolemma to become fragile and prone to repeated microinjuries. These microscopic tears accumulate over time, leading to irreversible damage and the eventual death of muscle fibers. This ongoing cycle of muscle fiber damage and death is a primary driver of muscle wasting in DMD.
As muscle fibers are lost, they are gradually replaced by fibrotic tissue and fat, which do not contribute to muscle function. This replacement process further exacerbates muscle weakness and reduces overall muscle mass. The progressive nature of this degeneration means that symptoms of DMD worsen over time, typically beginning in early childhood with difficulties in motor milestones such as walking and standing. By adolescence, many affected individuals lose the ability to walk independently, and the weakening of respiratory and cardiac muscles becomes a significant concern, often leading to life-limiting complications.
The genetic basis of DMD underscores the importance of dystrophin in muscle health, and its absence highlights the cascading effects of a single protein deficiency on the entire musculoskeletal system. Research into DMD has focused on understanding how dystrophin mutations lead to muscle fiber dysfunction, with the goal of developing targeted therapies. Approaches such as gene editing, exon skipping, and dystrophin replacement aim to restore dystrophin production or function, thereby slowing or halting the progression of muscle wasting. Despite these advancements, the complexity of the dystrophin gene and its mutations presents ongoing challenges in developing effective treatments.
In summary, genetic mutations in the dystrophin gene are the root cause of muscle wasting in DMD, as they disrupt the normal function of muscle fibers by eliminating or impairing dystrophin production. This disruption leads to repeated muscle fiber damage, necrosis, and eventual replacement with nonfunctional tissue, resulting in progressive weakness and atrophy. Understanding the molecular mechanisms behind dystrophin’s role in muscle stability is crucial for developing therapies that can mitigate the devastating effects of this disorder. Continued research and innovation are essential to improving outcomes for individuals affected by DMD.
Weak Muscles: Slower Reaction Times?
You may want to see also
Explore related products






![]()
Repeated muscle damage and repair cycles cause fibrosis, replacing functional muscle with non-contractile tissue
In Duchenne Muscular Dystrophy (DMD), repeated muscle damage and repair cycles play a central role in the progression of muscle wasting. DMD is caused by mutations in the dystrophin gene, leading to the absence or dysfunction of dystrophin protein, which is essential for maintaining muscle fiber integrity. Without dystrophin, muscle fibers become highly susceptible to mechanical stress during contraction, resulting in frequent damage. Each episode of damage triggers a repair process mediated by satellite cells, the resident stem cells of skeletal muscle. However, this repair mechanism is not flawless and becomes increasingly inefficient over time, contributing to the pathological changes observed in DMD.
The repair process in DMD muscles involves the activation, proliferation, and differentiation of satellite cells to replace damaged muscle fibers. Initially, this process can restore muscle function to some extent. However, the repetitive nature of muscle damage in DMD leads to chronic inflammation and the accumulation of fibro-adipogenic progenitors (FAPs). These FAPs, along with activated fibroblasts, secrete excessive amounts of extracellular matrix (ECM) components, such as collagen, as part of the repair response. Over time, this excessive ECM deposition results in fibrosis, the formation of scar-like tissue that replaces functional muscle fibers with non-contractile, fibrous material.
Fibrosis is a maladaptive consequence of the repeated damage and repair cycles in DMD. As fibrosis progresses, it disrupts the normal architecture of muscle tissue, impairing muscle contraction and force generation. The non-contractile fibrotic tissue also reduces muscle elasticity, making the remaining muscle fibers even more vulnerable to further damage. This creates a vicious cycle: increased fibrosis leads to greater muscle weakness, which in turn exacerbates mechanical stress and damage during movement. The progressive replacement of functional muscle with fibrotic tissue is a major driver of muscle wasting and functional decline in DMD.
Moreover, the fibrotic environment negatively impacts satellite cell function, further compromising the muscle’s ability to repair itself. Fibrosis alters the muscle niche, making it less conducive for satellite cell activation and differentiation. Additionally, the chronic inflammatory state associated with repeated damage and fibrosis releases factors that promote fibrosis over muscle regeneration. This shift from regenerative to fibrotic repair is a critical factor in the irreversible loss of muscle mass and function observed in DMD patients.
In summary, repeated muscle damage and repair cycles in DMD lead to fibrosis, a process where functional muscle tissue is replaced by non-contractile, fibrous material. This fibrosis is driven by chronic inflammation, excessive ECM deposition, and the accumulation of fibro-adipogenic progenitors. As fibrosis progresses, it impairs muscle function, reduces elasticity, and compromises the regenerative capacity of satellite cells, ultimately accelerating muscle wasting. Understanding this mechanism is crucial for developing targeted therapies aimed at breaking the cycle of damage, repair, and fibrosis in DMD.
Buspirone's Muscular Side Effects: Pain and Beyond
You may want to see also
Explore related products




![]()
Chronic inflammation accelerates muscle breakdown and impairs regeneration in DMD patients over time
Duchenne Muscular Dystrophy (DMD) is a devastating genetic disorder characterized by progressive muscle weakness and wasting due to the absence of dystrophin, a crucial protein for muscle fiber integrity. While the primary defect lies in the dystrophin gene, chronic inflammation plays a significant role in accelerating muscle breakdown and impairing regeneration over time. This inflammatory response, initially triggered by repeated cycles of muscle damage and repair, becomes chronic and contributes to the relentless progression of muscle wasting in DMD patients.
In healthy muscles, inflammation is a natural response to injury, facilitating the removal of damaged tissue and initiating repair processes. However, in DMD, the constant degeneration and regeneration of muscle fibers due to dystrophin deficiency lead to a persistent inflammatory state. Immune cells, particularly macrophages, infiltrate the muscle tissue and release pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-6. These cytokines not only promote further muscle damage but also create a hostile environment that hinders effective muscle regeneration. Over time, this chronic inflammation exacerbates muscle fiber necrosis, leading to irreversible muscle wasting.
Chronic inflammation in DMD also disrupts the balance between muscle protein synthesis and degradation. The elevated levels of pro-inflammatory cytokines activate proteolytic pathways, such as the ubiquitin-proteasome system and calpain, which degrade muscle proteins at an accelerated rate. Simultaneously, these cytokines suppress the activity of satellite cells, the resident stem cells responsible for muscle repair. Satellite cells are crucial for regenerating damaged muscle fibers, but in the presence of chronic inflammation, their proliferation and differentiation are impaired. This dual effect of increased protein breakdown and reduced regenerative capacity significantly contributes to the progressive muscle wasting observed in DMD patients.
Moreover, chronic inflammation in DMD promotes fibrosis, the excessive accumulation of connective tissue within muscle. Fibroblasts, activated by inflammatory signals, produce collagen and other extracellular matrix components, leading to the formation of scar tissue. This fibrotic tissue replaces functional muscle fibers, further diminishing muscle strength and function. The interplay between inflammation and fibrosis creates a vicious cycle, as fibrotic tissue itself can perpetuate inflammation, thereby accelerating muscle breakdown and impairing regeneration even further.
In summary, chronic inflammation is a critical driver of muscle wasting in DMD, acting through multiple mechanisms to accelerate muscle breakdown and impair regeneration. By promoting proteolysis, inhibiting satellite cell function, and fostering fibrosis, chronic inflammation exacerbates the degenerative process initiated by dystrophin deficiency. Understanding the role of inflammation in DMD pathogenesis highlights the importance of targeting inflammatory pathways as a potential therapeutic strategy to slow disease progression and preserve muscle function in affected individuals.
Alcohol and Muscle Weakness: What's the Link?
You may want to see also
![]()
Lack of dystrophin protein increases muscle membrane fragility, causing frequent injury and degeneration
Duchenne Muscular Dystrophy (DMD) is a genetic disorder characterized by progressive muscle weakness and wasting. At the core of this condition is the absence or severe deficiency of dystrophin, a crucial protein that plays a vital role in maintaining the integrity of muscle fibers. Dystrophin acts as a structural stabilizer, linking the internal cytoskeleton of muscle cells to the extracellular matrix via the muscle membrane (sarcolemma). In healthy muscles, this linkage provides mechanical support, protecting the muscle fibers from damage during contraction and relaxation. However, in individuals with DMD, the lack of dystrophin disrupts this protective mechanism, leading to increased fragility of the muscle membrane.
The absence of dystrophin weakens the sarcolemma, making it more susceptible to mechanical stress during muscle activity. Normally, dystrophin helps absorb and distribute the forces generated during muscle contraction, preventing tears or ruptures in the membrane. Without dystrophin, the sarcolemma becomes highly vulnerable to even minor mechanical strain. This fragility results in frequent microinjuries to the muscle fibers, which, over time, accumulate and lead to irreversible damage. Each injury triggers an inflammatory response, but the repetitive nature of these injuries overwhelms the muscle's repair mechanisms, causing a cycle of degeneration and attempted regeneration.
As muscle fibers repeatedly undergo injury and repair, they become increasingly compromised. The continuous breakdown of muscle tissue outpaces its ability to regenerate effectively, leading to the replacement of functional muscle tissue with fibrotic scar tissue and adipose (fat) tissue. This process, known as fibrosis, further weakens the muscle and contributes to the progressive loss of muscle mass and function observed in DMD. The degeneration of muscle fibers is not only localized but also systemic, affecting skeletal, cardiac, and respiratory muscles, which explains the widespread symptoms and complications associated with the disease.
The frequent injuries caused by dystrophin deficiency also lead to an influx of calcium ions into the muscle cells. Normally, the sarcolemma tightly regulates calcium levels, but its fragility allows excessive calcium entry, which activates harmful enzymes like calpains and triggers apoptosis (programmed cell death). This calcium-mediated damage exacerbates muscle fiber degeneration, accelerating the progression of muscle wasting. Additionally, the chronic inflammation associated with repeated injuries creates a toxic environment that further impairs muscle regeneration and promotes tissue breakdown.
In summary, the lack of dystrophin protein in DMD directly increases muscle membrane fragility, making it highly prone to injury during normal muscle use. These frequent injuries initiate a cascade of detrimental events, including inflammation, fibrosis, calcium-induced damage, and cell death, all of which contribute to progressive muscle degeneration and wasting. Understanding this mechanism underscores the critical role of dystrophin in muscle health and highlights the need for therapeutic strategies aimed at restoring its function or mitigating its absence in DMD.
Inflammation and Muscle Weakness: Is There a Link?
You may want to see also
![]()
Oxidative stress and mitochondrial dysfunction contribute to muscle cell death and atrophy in DMD
Duchenne Muscular Dystrophy (DMD) is a severe genetic disorder characterized by progressive muscle weakness and wasting due to the absence of dystrophin, a protein crucial for muscle fiber integrity. While the primary cause of muscle degeneration in DMD is the mechanical vulnerability of dystrophin-deficient muscle fibers, emerging evidence highlights oxidative stress and mitochondrial dysfunction as significant contributors to muscle cell death and atrophy in this condition. Oxidative stress occurs when there is an imbalance between the production of reactive oxygen species (ROS) and the antioxidant defense mechanisms in cells. In DMD, the repeated cycles of muscle damage and repair lead to chronic inflammation, which increases ROS production. This elevated oxidative stress directly damages cellular components, including lipids, proteins, and DNA, compromising muscle cell function and viability.
Mitochondria, often referred to as the "powerhouses" of the cell, play a central role in energy production and are particularly susceptible to oxidative damage. In DMD, mitochondrial dysfunction exacerbates muscle wasting by impairing ATP production, which is essential for muscle contraction and repair. Dystrophin deficiency disrupts the sarcolemma, leading to increased calcium influx into muscle cells. Excessive calcium levels further stress mitochondria, triggering the opening of the mitochondrial permeability transition pore (mPTP), which can result in mitochondrial swelling, depolarization, and cell death via apoptosis or necrosis. This vicious cycle of mitochondrial dysfunction and oxidative stress accelerates muscle fiber degeneration and contributes to the progressive atrophy observed in DMD.
Moreover, oxidative stress and mitochondrial dysfunction are interconnected in DMD, creating a feedback loop that amplifies muscle damage. Damaged mitochondria produce more ROS, which in turn exacerbates oxidative stress, leading to further mitochondrial impairment. This process is compounded by the reduced expression of antioxidant enzymes, such as superoxide dismutase (SOD) and catalase, in dystrophic muscles, leaving them more vulnerable to ROS-induced damage. Studies have shown that dystrophin deficiency alters the localization and function of mitochondrial proteins, disrupting their ability to maintain cellular homeostasis and repair oxidative damage.
Therapeutic strategies targeting oxidative stress and mitochondrial dysfunction hold promise for mitigating muscle wasting in DMD. Antioxidant supplementation, such as with vitamin E or N-acetylcysteine, has been explored to counteract ROS-induced damage. Additionally, compounds that enhance mitochondrial function, like coenzyme Q10 or mitochondrial-targeted peptides, have shown potential in preclinical models. Gene therapy approaches aimed at restoring dystrophin expression or modulating oxidative stress pathways are also under investigation. By addressing these underlying mechanisms, it may be possible to slow the progression of muscle atrophy and improve the quality of life for individuals with DMD.
In conclusion, oxidative stress and mitochondrial dysfunction are critical factors in the pathogenesis of muscle wasting in DMD, acting in conjunction with the primary dystrophin deficiency to accelerate muscle cell death and atrophy. Understanding the intricate relationship between these processes provides valuable insights into potential therapeutic targets. Future research should focus on developing interventions that effectively mitigate oxidative stress and enhance mitochondrial resilience, offering hope for more comprehensive treatment strategies in DMD.
Strained Muscles and Tingling: Unraveling the Connection and Symptoms
You may want to see also
Frequently asked questions
The primary cause of muscle wasting in DMD is the lack of dystrophin, a protein essential for muscle fiber stability. Without dystrophin, muscle fibers become vulnerable to damage during contraction, leading to repeated cycles of degeneration and regeneration, which eventually result in muscle wasting.
Inflammation plays a significant role in muscle wasting in DMD. As muscle fibers are damaged due to dystrophin deficiency, the body’s immune system responds with chronic inflammation. Over time, this inflammation leads to the accumulation of fibrotic tissue and fat, replacing functional muscle tissue and accelerating muscle wasting.
Yes, physical inactivity can exacerbate muscle wasting in DMD. While excessive strain should be avoided, moderate physical activity helps maintain muscle strength and function. Prolonged inactivity leads to disuse atrophy, compounding the muscle wasting caused by dystrophin deficiency. Balanced, supervised exercise is often recommended to slow progression.