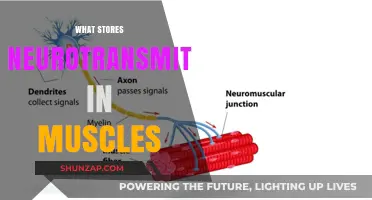
Spinal muscular atrophy (SMA) is a genetic disorder affecting the motor neurons that control voluntary muscle movement. These cells are located in the spinal cord, and when they cannot respond to signals from the nerves, the muscles atrophy – weakening and shrinking due to inactivity. SMA is one of the most prevalent genetic disorders affecting young children and is a major cause of death in infancy. SMA can manifest at any age, but the later the symptoms develop, the better the prognosis. While there is currently no cure for SMA, treatments such as physical therapy, occupational therapy, and medication can help manage symptoms and improve quality of life.
| Characteristics | Values |
|---|---|
| Disorder Type | Motor neuron disease |
| Affected Cells | Motor neurons |
| Cell Location | Spinal cord |
| Disorder Impact | Weakening and shrinking of muscles |
| Disorder Cause | Insufficient levels of SMN protein |
| Disorder Symptoms | Muscle weakness, trouble breathing, coughing, swallowing, tremors, scoliosis, decreased tendon reflexes |
| Disorder Diagnosis | Genetic testing, electromyography, muscle biopsy |
| Disorder Treatment | Physical therapy, occupational therapy, rehabilitation, stretching, strengthening exercises, speech therapy, assistive devices, nutritional support, feeding tube, medicines, nusinersen, onasemnogene abeparvovec |
| Disorder Severity | Varies with age of onset, types 0/1 most severe, type 4 least severe |
| Disorder Prevalence | 1 in 4,000 to 1 in 16,000 births worldwide |
Explore related products






![]()
Types of SMA
Spinal muscular atrophy (SMA) is a group of genetic diseases that affect the central nervous system, peripheral nervous system, and skeletal muscle movement. SMA is caused by a mutation in the SMN1 gene, which results in insufficient production of the SMN protein. This protein is crucial for the survival and functioning of motor neurons. The severity of SMA symptoms depends on the number of copies of the SMN2 gene, which can partially compensate for the loss of function of SMN1.
There are five types of SMA, categorized by the disease's severity and the age when symptoms begin:
Type 0 SMA or Prenatal SMA
Type 0 SMA is a rare and severe form of the disease that affects babies before they are born. It is characterized by decreased fetal movement in the weeks before delivery. Infants with Type 0 SMA experience severe muscle weakness and respiratory failure at birth, and typically only survive a few months.
Type 1 SMA or Infantile-Onset SMA
Type 1 SMA, also known as Werdnig-Hoffmann disease, is the most common and severe form of SMA. It occurs between birth and six months of age. Babies with Type 1 SMA have a characteristic bell-shaped body, with a narrow chest and large belly. They exhibit limited movement, lack head control, and often have severe breathing and feeding difficulties. Without treatment, children with Type 1 SMA rarely survive beyond two years of age.
Type II SMA
Type II SMA typically appears late in infancy, usually between seven and eighteen months of age. It progresses more slowly than Type I SMA. Infants with Type II SMA can typically suck and swallow, and do not initially have serious breathing problems. They may be able to sit independently but will likely become confined to a wheelchair over time due to progressive muscle weakness.
Type III SMA or Juvenile SMA
Type III SMA, also known as Kugelberg-Welander syndrome, usually affects children between 18 months and three years of age, although it can sometimes go undiagnosed until adolescence. Children with Type III SMA can walk initially but will eventually lose this ability and require a wheelchair. They experience muscle weakness, particularly in the legs, and may fall frequently. Type III SMA is the mildest form of SMA in children.
Type IV SMA or Late-Onset SMA
Type IV SMA is the rarest form of SMA, affecting less than 1% of patients. It typically begins after age 35 and causes mild motor impairment. Most individuals with Type IV SMA remain mobile into their 60s, after which they may require walking aids.
Mastering Muscle Memory: The Ultimate Guide to OIA Memorization
You may want to see also
Explore related products






![]()
Causes of SMA
Spinal muscular atrophy (SMA) is a genetic disorder affecting the central nervous system, peripheral nervous system, and voluntary muscle movement (skeletal muscle). It is caused by insufficient levels of the survival motor neuron (SMN) protein, which maintains the health and normal function of motor neurons. The most common form of SMA is caused by mutations of the survival motor neuron 1 gene (SMN1) on the fifth chromosome, resulting in insufficient expression levels of the SMN protein.
SMN is essential to normal motor function because it enables muscles to receive signals from nerves. People with SMA have insufficient levels of SMN protein, which leads to the loss of motor neurons in the spinal cord and causes weakness and wasting of the skeletal muscles. The muscles most affected are those closest to the center of the body, such as the shoulders, hips, thighs, and upper back. The lower limbs are typically more affected than the upper limbs, and deep tendon reflexes are decreased.
The SMN1 gene is typically passed down from both parents, with the child inheriting one mutated copy from each parent. Those with only one mutated copy are carriers of the disease but do not exhibit any symptoms. In rare cases, SMA can also be caused by changes in other genes, such as the UBE1 gene on the X chromosome, the DYNC1H1 gene on chromosome 14, and the IGHMBP2 gene on chromosome 11.
SMA is classified into types 1 through 4 based on the age of onset and physical milestones achieved. Type 1 SMA, also known as Werdnig-Hoffman disease, is the most severe form, striking infants within the first six months of life. Type 2 SMA is intermediate, affecting children between six and 18 months. Type 3 SMA, or juvenile SMA, emerges in children 18 months or older and can become evident as late as the teenage years. Type 4 SMA develops in adulthood and rarely impacts lifespan or causes severe disability.
Muscle Milk's Creatine Content: What You Need to Know
You may want to see also
Explore related products






![]()
Symptoms of SMA
Spinal muscular atrophy (SMA) is a genetic disorder that affects the motor neurons—nerve cells that control voluntary muscle movement. These cells are located in the spinal cord. The symptoms of SMA vary, and may be mild or disabling, but they involve a weakness of the muscles that control movement. The muscles closest to the center of the body, such as those of the shoulders, hips, thighs, and upper back, are the most affected. The lower limbs are more impacted than the upper limbs, and deep tendon reflexes are decreased.
SMA is classified into types 1 through 4, depending on the age of onset. Type 1 SMA, also known as Werdnig-Hoffmann disease or infantile-onset SMA, is the most common and severe form, affecting about 60% of babies born with SMA. Symptoms typically appear within the first six months of life and include severe muscle weakness, trouble breathing, coughing, and swallowing. Babies with type 1 SMA may be unable to sit up or raise their head due to weak core and neck muscles. They often require breathing assistance or a feeding tube and may not survive past their second birthday without aggressive therapy.
Type 2 SMA, also known as intermediate SMA or Dubowitz disease, affects about 30% of infants with SMA. Symptoms typically appear between six and 18 months of age. Children with type 2 SMA can sit independently but have difficulty standing and walking and swallowing. They experience muscle weakness, particularly in the lower limbs, and are prone to scoliosis and tremors in the fingers or tongue. Respiratory challenges can shorten their lives as the disease progresses.
Type 3 SMA, also known as juvenile SMA or Kugelberg-Welander syndrome, accounts for about 10% of infants born with SMA. Symptoms may emerge as early as 18 months or as late as the teenage years. Individuals with type 3 SMA can usually walk and stand for limited periods, especially early in the course of the illness. They may experience difficulty with tasks such as running, rising from a chair, or climbing stairs.
Type 4 SMA, also known as late-onset SMA, is the rarest form, occurring in less than 1% of people with SMA. Onset typically occurs in adulthood, most commonly after 35 years of age. Symptoms are generally milder and include gradual muscle weakness in the arms and legs, affecting mobility and daily tasks over time. Individuals with type 4 SMA may experience fatigue, muscle aches, cramps, or numbness. They typically have a normal life expectancy.
Severed Muscle: What to Do and Recovery Timeline
You may want to see also
Explore related products


![sma 13-4056 TIP 44 Substance Abuse Treatment for Adults in the Criminal Justice System, SMA 13-4056, 2013 [Loose leaf]](https://m.media-amazon.com/images/I/71R2x4Z77eL._AC_UL320_.jpg)
![Addressing the Specific Needs of Women - Substance Abuse Treatment SMA 15-4426 TIP 51 - A TREATMENT IMPROVEMENT PROTOCOL [Black and White Loose Leaf Edition]](https://m.media-amazon.com/images/I/71z8ZXOcr+L._AC_UL320_.jpg)
![Integrating Substance Abuse Treatment and Vocational Services TIP 38 SMA 12-4216 2018 - A TREATMENT IMPROVEMENT PROTOCOL [Black and White Loose Leaf Edition]](https://m.media-amazon.com/images/I/916aMbpgqUL._AC_UL320_.jpg)

![]()
Diagnosis of SMA
Spinal muscular atrophy (SMA) is a genetic disorder that affects the motor neurons—nerve cells that control voluntary muscle movement. SMA is diagnosed after symptoms start and after testing is done. The symptoms of SMA are similar to those of several other diseases affecting the muscles, so doctors must conduct tests to determine whether a patient's muscle atrophy is due to SMA.
The first steps in diagnosing SMA usually involve an in-office physical examination, a neurological exam, and a review of the patient's family history. Doctors will look for signs such as muscle weakness, a history of motor difficulties, loss of motor skills, proximal muscle weakness, and tongue fasciculations (involuntary twitches). A blood test for creatine kinase (CK) may also be ordered, as high levels of this enzyme indicate that muscle damage has occurred.
If SMA is suspected, doctors will likely recommend genetic testing, which is currently the least invasive and most accurate way to diagnose SMA. This involves taking a blood sample to look for mutations or deletions of the SMN1 gene. This test identifies at least 95% of SMA Types I, II, and III and may reveal if a person is a carrier for SMA.
Other diagnostic tests for SMA include electromyography (EMG), which records the electrical activity of the muscles during contraction and at rest, and nerve conduction velocity studies, which measure the nerve's ability to send an electrical signal. In rare cases, a muscle biopsy may be performed, involving the removal of a small piece of muscle tissue for examination under a microscope.
Building Lean Muscle: Understanding the Dry Lean Muscle Concept
You may want to see also
Explore related products
![SMA 12-4078 - TIP 29 - Substance Use Disorder Treatment for People With Physical and Cognitive Disabilities [Loose Leaf Edition]](https://m.media-amazon.com/images/I/81yC1ImysdL._AC_UL320_.jpg)





![]()
Treatment of SMA
Spinal muscular atrophy (SMA) is a rare neuromuscular disease, with an estimated incidence of about 1 in 10,000 live births. It is caused by a genetic mutation in the SMN1 gene, which results in insufficient production of the survival motor neuron (SMN) protein. This protein is crucial for the survival and function of motor neurons, and its deficiency leads to the degeneration and death of these neurons. SMA affects the central nervous system, peripheral nervous system, and voluntary muscle movement, causing muscle weakness and atrophy.
While there is currently no cure for SMA, several treatments are available to manage the condition and improve patients' quality of life. Treatment options primarily focus on increasing SMN protein levels and targeting other affected systems, pathways, and processes. Here is an overview of the treatment approaches for SMA:
SMN-based Approaches:
SMN-based approaches aim to increase the levels of SMN protein in the body. This can be achieved through various therapies and medications. Gene replacement therapy, also known as onasemnogene abeparvovec -xioi (brand name Zolgensma), is a treatment option for children with SMA. Zolgensma was approved by the FDA in 2019 for children under 2 years old with all forms and types of SMA. It is the only FDA-approved gene therapy for SMA as of 2023.
Other SMN-based medications include nusinersen (Spinraza) and risdiplam (Evrysdia), which are used to increase the production of SMN protein. These medications are approved for the treatment of SMA in both children and adults.
Non-SMN Approaches:
Non-SMN approaches target other systems and pathways affected by SMA, such as muscles or nerves. Researchers believe that a combination of SMN-based and non-SMN treatments may provide the most benefit to patients with SMA. This could involve taking two drugs together or switching between different treatments at various stages of the disease.
Early Intervention:
Regardless of the treatment approach, it is crucial to begin therapy as soon as possible after diagnosis. Early intervention is especially critical for SMN-enhancing therapies to prevent or slow down motor neuron loss. Clinical trials have shown that infants and children who started SMA treatment earlier had better outcomes than those who started later. Therefore, prompt diagnosis and treatment are essential for optimizing the effectiveness of available therapies.
In summary, while there is no cure for SMA, approved treatments, including gene therapy and medications, can help manage the condition and improve patients' outcomes. Early diagnosis and timely initiation of therapy are vital to maximizing the benefits of these treatments. As research progresses, there is ongoing hope for the development of additional effective treatment options for SMA.
Exploring the Anatomy of Fascia and Fascicle Interactions
You may want to see also