A decrease in oxygen (O₂) levels, a condition known as hypoxia, triggers smooth muscle relaxation through several interconnected mechanisms. Hypoxia activates specific signaling pathways, such as the upregulation of hypoxia-inducible factors (HIFs), which modulate gene expression to enhance cellular survival under low-oxygen conditions. Additionally, hypoxia reduces the availability of O₂ for oxidative phosphorylation, leading to a shift toward anaerobic metabolism and decreased ATP production. This energy deficit impairs the ability of smooth muscle cells to maintain calcium (Ca²⁺) homeostasis, resulting in reduced Ca²⁰ influx and decreased activation of contractile proteins like myosin light chain kinase (MLCK). Furthermore, hypoxia promotes the release of vasodilatory mediators, such as nitric oxide (NO) and adenosine, which further contribute to smooth muscle relaxation. Collectively, these processes explain why a decrease in O₂ levels induces smooth muscle relaxation, a response observed in various physiological and pathological contexts, including vascular regulation and hypoxia-induced tissue adaptation.
| Characteristics | Values |
|---|---|
| Mechanism | Decreased O2 levels lead to hypoxia, which activates specific signaling pathways in smooth muscle cells. |
| Key Pathway | Hypoxia induces the activation of potassium (K+) channels, particularly ATP-sensitive K+ (KATP) channels, leading to hyperpolarization of the cell membrane. |
| Hyperpolarization Effect | Hyperpolarization reduces the influx of calcium (Ca2+) ions into the cell, decreasing intracellular Ca2+ concentration. |
| Calcium Role | Lower intracellular Ca2+ levels result in decreased activation of the contractile machinery (e.g., calmodulin and myosin light chain kinase), leading to smooth muscle relaxation. |
| Additional Factors | Hypoxia also increases the production of nitric oxide (NO) and adenosine, which further contribute to vasodilation and smooth muscle relaxation. |
| Tissue Specificity | This response is particularly prominent in vascular smooth muscle, where relaxation leads to vasodilation, improving blood flow to hypoxic tissues. |
| Clinical Relevance | This mechanism is crucial in conditions like ischemia, where decreased O2 triggers vasodilation to enhance oxygen delivery to affected tissues. |
| Energy Metabolism | Hypoxia shifts cellular metabolism toward anaerobic pathways, reducing ATP production, which indirectly supports KATP channel activation and relaxation. |
| Reversibility | The relaxation is reversible; restoring O2 levels normalizes intracellular Ca2+ and restores smooth muscle tone. |
Explore related products






What You'll Learn
- Role of Hypoxia-Inducible Factors (HIFs) in Smooth Muscle Relaxation
- Impact of Reduced ATP Production on Calcium Signaling Pathways
- Effect of Nitric Oxide (NO) Release in Low Oxygen Conditions
- Changes in Potassium Channel Activity During Hypoxia
- Influence of Reactive Oxygen Species (ROS) on Smooth Muscle Tone
![]()
Role of Hypoxia-Inducible Factors (HIFs) in Smooth Muscle Relaxation
Under conditions of decreased oxygen availability (hypoxia), smooth muscle relaxation occurs as a physiological response to ensure adequate blood flow to vital organs. This process is intricately regulated by Hypoxia-Inducible Factors (HIFs), a family of transcription factors that play a pivotal role in cellular adaptation to low oxygen levels. HIFs are central to understanding why a decrease in O₂ triggers smooth muscle relaxation, as they orchestrate a cascade of molecular events that modulate vascular tone and tissue perfusion.
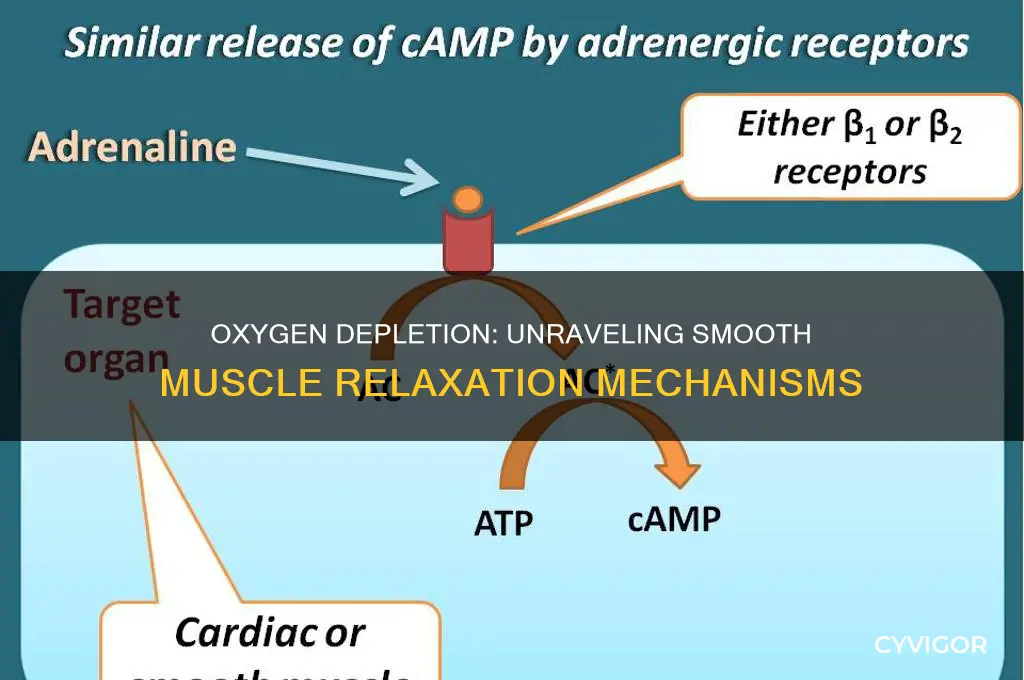
HIFs consist of two subunits: HIF-1α (or HIF-2α) and HIF-1β. Under normoxic conditions, HIF-1α is continuously synthesized but rapidly degraded by prolyl hydroxylases (PHDs), which require oxygen as a co-substrate. However, during hypoxia, PHD activity is inhibited, allowing HIF-1α to stabilize, translocate to the nucleus, and dimerize with HIF-1β. This complex binds to hypoxia-response elements (HREs) in the promoters of target genes, activating their transcription. Among the genes regulated by HIFs are those involved in vasodilation, such as nitric oxide synthase (NOS) and vascular endothelial growth factor (VEGF), which contribute to smooth muscle relaxation.
One of the key mechanisms by which HIFs promote smooth muscle relaxation is through the upregulation of NOS, particularly endothelial NOS (eNOS). Increased eNOS activity enhances nitric oxide (NO) production, a potent vasodilator that activates soluble guanylate cyclase (sGC) in smooth muscle cells. This leads to elevated cyclic guanosine monophosphate (cGMP) levels, which in turn activate protein kinase G (PKG). PKG phosphorylates target proteins, including calcium channels and myosin light chain phosphatase, resulting in reduced intracellular calcium levels and decreased smooth muscle contraction, thereby inducing relaxation.
HIFs also stimulate the expression of VEGF, which acts indirectly to promote smooth muscle relaxation. VEGF enhances vascular permeability and angiogenesis, improving tissue oxygenation. Additionally, VEGF can induce the production of other vasodilatory factors, such as prostacyclin and endothelium-derived hyperpolarizing factors (EDHFs), which further contribute to smooth muscle relaxation. This multifaceted approach ensures that tissues receive adequate oxygen supply during hypoxic conditions.
Furthermore, HIFs modulate the expression of potassium channels in smooth muscle cells, such as calcium-activated potassium channels (KCa) and ATP-sensitive potassium channels (KATP). Activation of these channels leads to hyperpolarization of the cell membrane, reducing calcium influx and promoting relaxation. HIF-mediated upregulation of these channels is a critical component of the hypoxic response, providing an additional layer of regulation to ensure vascular tone is appropriately adjusted.
In summary, Hypoxia-Inducible Factors (HIFs) are essential mediators of smooth muscle relaxation during hypoxia. By stabilizing under low oxygen conditions, HIFs activate the transcription of genes involved in vasodilation, such as NOS and VEGF, while also modulating potassium channel expression. These actions collectively reduce intracellular calcium levels and promote smooth muscle relaxation, ensuring optimal tissue perfusion in response to decreased O₂ availability. Understanding the role of HIFs in this process provides valuable insights into the physiological mechanisms of vascular regulation and potential therapeutic targets for hypoxia-related disorders.
Small Fiber Neuropathy and Muscle Twitching: Understanding the Connection
You may want to see also
Explore related products






![]()
Impact of Reduced ATP Production on Calcium Signaling Pathways
A decrease in oxygen (O₂) availability, such as during hypoxia, significantly impacts cellular energy production, primarily by reducing adenosine triphosphate (ATP) synthesis via mitochondrial oxidative phosphorylation. This reduction in ATP has profound effects on calcium signaling pathways, which are critical for smooth muscle contraction and relaxation. Calcium signaling in smooth muscle cells relies heavily on ATP-dependent processes, including the activity of plasma membrane calcium channels, sarcoplasmic reticulum (SR) calcium pumps, and calcium-binding proteins. When ATP production decreases, these processes are compromised, leading to alterations in intracellular calcium concentrations ([Ca²⁺]i) and subsequent smooth muscle relaxation.
One of the key impacts of reduced ATP production is the impairment of the sarco/endoplasmic reticulum Ca²⁺-ATPase (SERCA) pump. The SERCA pump is responsible for actively transporting Ca²⁺ from the cytosol into the SR, thereby lowering [Ca²⁺]i and promoting muscle relaxation. This pump is highly ATP-dependent, and its activity diminishes when ATP levels decline. As a result, Ca²⁺ reuptake into the SR is reduced, leading to elevated [Ca²⁺]i. However, paradoxically, prolonged hypoxia and ATP depletion also impair the release of Ca²⁺ from the SR via ryanodine receptors (RyRs), which are crucial for calcium-induced calcium release (CICR) during muscle contraction. The combined effect of reduced SERCA activity and RyR dysfunction leads to a net decrease in [Ca²⁺]i available for contraction, promoting smooth muscle relaxation.
Another critical aspect of calcium signaling affected by reduced ATP production is the function of plasma membrane calcium channels, such as voltage-gated calcium channels (VGCCs) and store-operated calcium channels (SOCs). VGCCs, which mediate Ca²⁺ influx in response to membrane depolarization, require ATP for proper function and regulation. Hypoxia-induced ATP depletion can lead to decreased VGCC activity, reducing Ca²⁺ entry into the cell. Similarly, SOCs, which are activated by SR calcium depletion, rely on ATP for associated signaling molecules like stromal interaction molecule 1 (STIM1) and Orai1. ATP depletion disrupts this signaling, further limiting Ca²⁺ influx. Both mechanisms contribute to reduced [Ca²⁺]i and smooth muscle relaxation.
Additionally, ATP depletion affects the activity of calcium-binding proteins such as calmodulin and troponin, which are essential for translating calcium signals into muscle contraction. Calmodulin, for instance, requires ATP for its interaction with target proteins, including myosin light-chain kinase (MLCK), which phosphorylates myosin light chains to initiate contraction. When ATP is scarce, calmodulin-dependent pathways are impaired, reducing MLCK activity and decreasing the sensitivity of contractile machinery to calcium. This desensitization further contributes to smooth muscle relaxation.
In summary, reduced ATP production due to hypoxia disrupts multiple components of calcium signaling pathways in smooth muscle cells. Impaired SERCA pump activity, dysfunctional RyRs, reduced plasma membrane calcium channel function, and compromised calcium-binding protein activity collectively lead to decreased [Ca²⁺]i and diminished contractile capability. These mechanisms explain why a decrease in O₂, by reducing ATP availability, ultimately causes smooth muscle relaxation. Understanding these pathways provides insights into the physiological responses to hypoxia and potential therapeutic targets for conditions involving impaired oxygen supply.
Ozempic's Muscle Loss: What's the Deal?
You may want to see also
Explore related products






![]()
Effect of Nitric Oxide (NO) Release in Low Oxygen Conditions
In low oxygen conditions, also known as hypoxia, the release of nitric oxide (NO) plays a crucial role in mediating smooth muscle relaxation. This process is particularly important in vascular smooth muscle, where it helps regulate blood flow and oxygen delivery to tissues. When oxygen levels decrease, cells respond by activating specific signaling pathways that lead to the production of NO. This gasotransmitter is synthesized by the enzyme nitric oxide synthase (NOS), which becomes upregulated under hypoxic conditions. The increased NO production is a protective mechanism aimed at dilating blood vessels, thereby enhancing oxygen supply to hypoxic tissues.
The effect of NO release in low oxygen conditions is primarily mediated through its interaction with soluble guanylate cyclase (sGC) in smooth muscle cells. Upon binding to sGC, NO stimulates the conversion of guanosine triphosphate (GTP) to cyclic guanosine monophosphate (cGMP). Elevated cGMP levels activate protein kinase G (PKG), which initiates a cascade of events leading to smooth muscle relaxation. Specifically, PKG phosphorylates target proteins such as myosin light chain phosphatase, reducing the phosphorylation of myosin light chains and decreasing the contractile force in smooth muscle cells. This mechanism is fundamental to understanding why a decrease in O2 causes smooth muscle relaxation.
Hypoxia-induced NO release also involves the stabilization of hypoxia-inducible factor-1α (HIF-1α), a key transcription factor that regulates the cellular response to low oxygen. Under normoxic conditions, HIF-1α is rapidly degraded, but hypoxia inhibits this process, allowing HIF-1α to accumulate and translocate to the nucleus. There, it promotes the transcription of genes encoding NOS isoforms, particularly endothelial NOS (eNOS), thereby increasing NO production. This HIF-1α-mediated upregulation of NOS is a critical link between hypoxia and NO-dependent smooth muscle relaxation, ensuring that tissues can adapt to reduced oxygen availability.
Another important aspect of NO release in low oxygen conditions is its role in mitigating oxidative stress. Hypoxia can lead to the generation of reactive oxygen species (ROS), which may impair vascular function. NO acts as an antioxidant by reacting with superoxide radicals to form peroxynitrite, thus reducing oxidative damage. Additionally, NO enhances blood flow, which helps remove waste products and delivers antioxidants to hypoxic tissues. This dual action of NO—both as a vasodilator and an antioxidant—is essential for maintaining vascular homeostasis under hypoxic conditions.
In summary, the release of nitric oxide in low oxygen conditions is a vital adaptive response that promotes smooth muscle relaxation through cGMP-PKG signaling, HIF-1α-mediated NOS upregulation, and antioxidant effects. This mechanism ensures that blood vessels dilate, improving oxygen delivery to hypoxic tissues and preserving cellular function. Understanding the effect of NO release in hypoxia provides valuable insights into the physiological responses to reduced oxygen levels and highlights the importance of NO as a key mediator in vascular regulation.
Allergies and Muscle Weakness: Is There a Link?
You may want to see also
Explore related products






![]()
Changes in Potassium Channel Activity During Hypoxia
During hypoxia, a decrease in oxygen (O₂) levels triggers a cascade of cellular responses that influence smooth muscle relaxation. One critical mechanism involves changes in potassium (K⁺) channel activity. Potassium channels play a pivotal role in regulating membrane potential, and their modulation during hypoxia is central to understanding smooth muscle relaxation. Hypoxia-induced alterations in K⁺ channel activity lead to hyperpolarization of the cell membrane, which reduces the likelihood of voltage-gated calcium (Ca²⁺) channels opening. This decrease in intracellular Ca²⁺ concentration results in smooth muscle relaxation, as Ca²⁺ is essential for muscle contraction.
Among the K⁺ channels, ATP-sensitive K⁺ (KATP) channels are particularly significant during hypoxia. Under normal oxygen conditions, these channels are closed due to sufficient ATP levels. However, during hypoxia, ATP production decreases, leading to an increase in ADP and a drop in the ATP/ADP ratio. This change activates KATP channels, allowing K⁺ efflux and subsequent hyperpolarization of the cell membrane. The hyperpolarized state inhibits Ca²⁺ influx, thereby promoting smooth muscle relaxation. This mechanism is especially prominent in vascular smooth muscle, where KATP channel activation contributes to vasodilation in response to hypoxia.
Another class of K⁺ channels affected by hypoxia is the voltage-gated K⁺ (Kv) channels. Hypoxia can enhance the activity of certain Kv channel subtypes, further contributing to membrane hyperpolarization. This upregulation is often mediated by signaling pathways activated under low O₂ conditions, such as those involving hypoxia-inducible factor-1 (HIF-1). Increased Kv channel activity reinforces the reduction in Ca²⁺ entry, amplifying the relaxation response in smooth muscle cells. The interplay between KATP and Kv channels during hypoxia highlights the complexity of K⁺ channel regulation in this context.
Additionally, hypoxia can modulate the activity of inwardly rectifying K⁺ (Kir) channels, which are crucial for maintaining resting membrane potential. Under hypoxic conditions, Kir channel activity may increase, facilitating K⁺ efflux and hyperpolarization. This effect is particularly relevant in airway and vascular smooth muscles, where Kir channels contribute to hypoxia-induced relaxation. The coordinated activation of Kir, KATP, and Kv channels ensures a robust hyperpolarization response, effectively counteracting the contractile effects of Ca²⁺.
In summary, changes in potassium channel activity during hypoxia are a key driver of smooth muscle relaxation. Activation of KATP channels due to decreased ATP levels, upregulation of Kv channels via hypoxia-induced signaling, and enhanced Kir channel activity collectively hyperpolarize the cell membrane. This hyperpolarization reduces Ca²⁺ influx, leading to relaxation. Understanding these mechanisms provides insights into how hypoxia modulates smooth muscle tone and highlights the critical role of K⁺ channels in this process.
Trunk Muscles Driving Waist Flexion: Key Players and Functions
You may want to see also
Explore related products






![]()
Influence of Reactive Oxygen Species (ROS) on Smooth Muscle Tone
The influence of Reactive Oxygen Species (ROS) on smooth muscle tone is a critical aspect of understanding why a decrease in O₂ levels can lead to smooth muscle relaxation. ROS, including superoxide anion (O₂⁻), hydrogen peroxide (H₂O₂), and hydroxyl radicals (OH⁻), are naturally produced during cellular respiration, particularly in the mitochondria. Under physiological conditions, ROS act as signaling molecules, regulating various cellular processes, including smooth muscle contraction and relaxation. However, their role becomes particularly significant in hypoxic conditions, where O₂ availability is reduced. In hypoxia, the balance between ROS production and scavenging is disrupted, leading to alterations in smooth muscle tone.
A decrease in O₂ levels triggers a cascade of events that influence ROS production and, consequently, smooth muscle function. Normally, adequate O₂ levels allow for efficient electron transport chain (ETC) activity in the mitochondria, minimizing electron leakage and ROS generation. However, during hypoxia, the ETC becomes less efficient, leading to increased electron leakage and elevated ROS production. This surge in ROS can activate specific signaling pathways, such as those involving Rho-kinase and protein kinase C (PKC), which are known to modulate smooth muscle contraction. Paradoxically, while ROS can promote contraction in some contexts, their excessive accumulation in hypoxia often leads to smooth muscle relaxation by disrupting calcium homeostasis and inhibiting contractile machinery.
One key mechanism by which ROS influence smooth muscle tone is through their interaction with calcium channels and signaling pathways. ROS can reduce calcium influx by inhibiting L-type calcium channels, which are essential for smooth muscle contraction. Additionally, ROS can activate calcium-sensitive pathways that promote calcium reuptake into the sarcoplasmic reticulum or extrusion from the cell, thereby lowering cytosolic calcium levels. Since calcium is a critical mediator of smooth muscle contraction, its reduction directly contributes to muscle relaxation. This effect is particularly pronounced in hypoxic conditions, where the imbalance in ROS production exacerbates calcium dysregulation.
Another important aspect of ROS influence on smooth muscle tone is their role in activating specific relaxation pathways. For instance, ROS can stimulate the production of nitric oxide (NO) by upregulating endothelial nitric oxide synthase (eNOS) or through direct reactions with superoxide to form peroxynitrite. NO is a potent vasodilator that promotes smooth muscle relaxation by activating soluble guanylate cyclase and increasing cyclic GMP levels, which in turn inhibit calcium influx and activate protein phosphatases. In hypoxia, the interplay between ROS and NO signaling becomes a critical determinant of smooth muscle tone, often tipping the balance toward relaxation.
Furthermore, ROS can modulate smooth muscle tone by affecting the cytoskeleton and contractile proteins. Excessive ROS can oxidize and inactivate key components of the contractile machinery, such as actin and myosin, thereby impairing muscle contraction. Additionally, ROS can activate pathways that promote the phosphorylation of myosin light chain phosphatase (MLCP), an enzyme that dephosphorylates myosin light chains and induces relaxation. These effects collectively contribute to the relaxation of smooth muscle observed in hypoxic conditions.
In summary, the influence of ROS on smooth muscle tone is multifaceted and closely tied to O₂ availability. A decrease in O₂ levels disrupts the balance of ROS production, leading to their excessive accumulation and activation of relaxation pathways. Through mechanisms involving calcium dysregulation, NO signaling, and modulation of contractile proteins, ROS play a pivotal role in mediating smooth muscle relaxation during hypoxia. Understanding these processes provides valuable insights into the physiological and pathological responses of smooth muscle to changes in O₂ levels.
Seizures and Muscle Pain: Understanding Post-Seizure Soreness Causes
You may want to see also
Frequently asked questions
A decrease in O2 (hypoxia) causes smooth muscle relaxation due to the activation of potassium channels, leading to hyperpolarization of the muscle cell membrane and reduced calcium influx, which decreases muscle contraction.
Hypoxia reduces intracellular calcium levels by inhibiting calcium release from the sarcoplasmic reticulum and decreasing calcium entry through voltage-gated calcium channels, resulting in muscle relaxation.
Potassium channels open in response to hypoxia, allowing potassium ions to exit the cell. This hyperpolarizes the membrane, making it less likely for voltage-gated calcium channels to open, thereby reducing calcium influx and promoting relaxation.
Signaling pathways involving hypoxia-inducible factors (HIFs), nitric oxide (NO), and adenosine are activated during hypoxia. These pathways contribute to the relaxation of smooth muscle by modulating ion channels and reducing contractile activity.
No, hypoxia-induced smooth muscle relaxation is more prominent in certain types of smooth muscle, such as those in blood vessels (vasodilation), but may not occur uniformly across all smooth muscle tissues, depending on their specific physiological roles and adaptations.