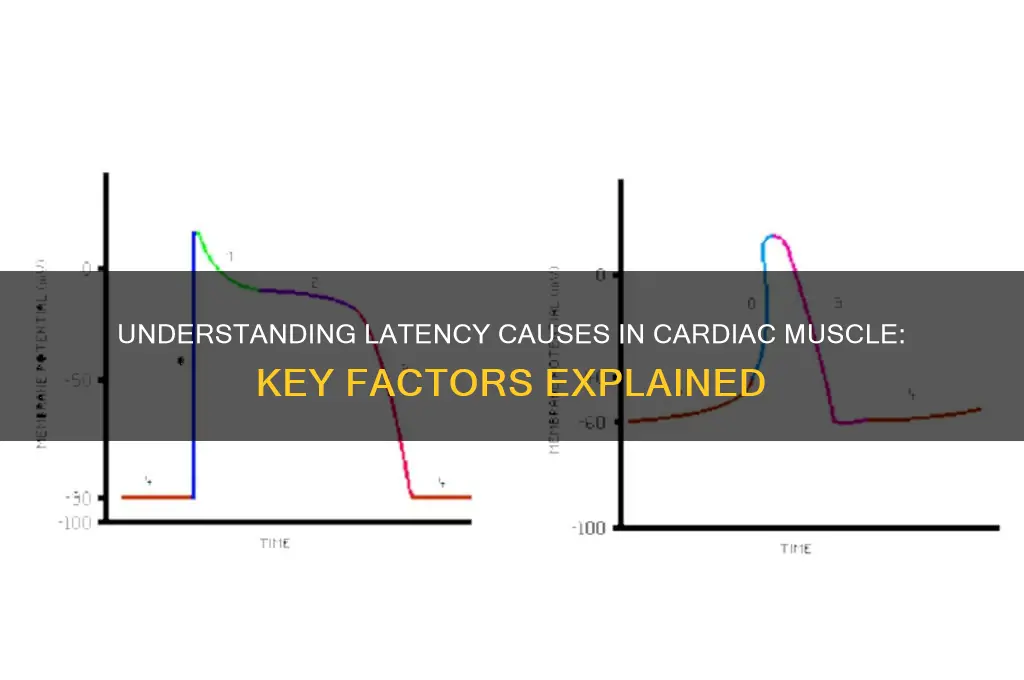
Latency in cardiac muscle refers to the delay between the initiation of an electrical stimulus and the subsequent mechanical contraction of the heart. This phenomenon is primarily caused by the complex interplay of electrophysiological and biochemical processes within cardiomyocytes. Key factors contributing to latency include the time required for depolarization to propagate through the sarcolemma and transverse tubules, the activation of voltage-gated calcium channels, and the subsequent release of calcium ions from the sarcoplasmic reticulum. Additionally, the interaction between calcium and troponin-C, which initiates the sliding filament mechanism, further contributes to the delay. External factors such as temperature, pH levels, and the presence of certain drugs or electrolytes can also influence latency by modulating these processes. Understanding these mechanisms is crucial for diagnosing and treating cardiac disorders associated with abnormal contraction timing.
Explore related products






What You'll Learn
- Slow Calcium Release: Delayed calcium release from sarcoplasmic reticulum slows muscle contraction, increasing latency
- Impaired Ion Channels: Dysfunctional ion channels disrupt electrical signaling, causing delayed depolarization and latency
- Reduced ATP Levels: Low energy availability slows cross-bridge cycling, prolonging contraction and relaxation times
- Hypoxia Effects: Oxygen deprivation impairs metabolic processes, slowing muscle fiber response and increasing latency
- Aging Impact: Age-related fibrosis and stiffening reduce cardiac muscle elasticity, delaying contraction and relaxation
![]()
Slow Calcium Release: Delayed calcium release from sarcoplasmic reticulum slows muscle contraction, increasing latency
In cardiac muscle, the rapid and coordinated release of calcium ions (Ca²⁺) from the sarcoplasmic reticulum (SR) is essential for initiating muscle contraction. This process, known as calcium-induced calcium release (CICR), triggers the interaction between actin and myosin filaments, leading to myocardial fiber shortening. However, when calcium release from the SR is delayed, it directly slows the onset of contraction, thereby increasing latency in cardiac muscle. This delay can occur due to impaired function of the ryanodine receptors (RyR2), the calcium release channels on the SR, or due to reduced SR calcium stores. Such impairments disrupt the synchrony of calcium release, causing a lag between electrical excitation (action potential) and mechanical response (contraction), which manifests as increased latency.
The mechanisms underlying slow calcium release often involve dysregulation of RyR2 channels. These channels can become leaky or desensitized due to factors like oxidative stress, phosphorylation abnormalities, or genetic mutations. When RyR2 function is compromised, the SR releases calcium ions more slowly or in smaller quantities, delaying the rise in cytosolic calcium concentration ([Ca²⁺]). This sluggish increase in [Ca²⁺] postpones the activation of troponin C, a protein critical for initiating actin-myosin cross-bridge formation. As a result, the time between electrical depolarization and the start of muscle contraction is prolonged, contributing to latency in cardiac muscle function.
Another factor contributing to slow calcium release is a depletion or imbalance in SR calcium stores. The SR relies on the sarcoplasmic reticulum ATPase (SERCA) pump to actively reuptake calcium ions into the SR lumen during diastole. If SERCA function is impaired, either due to reduced expression, post-translational modifications, or energy depletion (e.g., low ATP levels), the SR cannot adequately refill its calcium stores. This reduction in available calcium for release during systole leads to a delayed and diminished calcium transient, slowing contraction and increasing latency. Conditions like heart failure or ischemia often exacerbate SERCA dysfunction, further prolonging calcium release and contraction latency.
Additionally, extracellular factors such as altered calcium influx through L-type calcium channels in the sarcolemma can indirectly contribute to slow calcium release. During an action potential, L-type calcium channels open, allowing a small influx of Ca²⁺ that triggers CICR from the SR. If these channels are dysfunctional or blocked, the initial calcium spark is reduced, leading to a delayed or weakened SR calcium release. This cascade of events slows the overall contraction process, increasing latency. Pharmacological agents or pathological conditions affecting L-type calcium channels can thus indirectly contribute to delayed calcium release and subsequent latency in cardiac muscle.
In summary, slow calcium release from the sarcoplasmic reticulum is a significant cause of latency in cardiac muscle contraction. This delay arises from impaired RyR2 function, depleted SR calcium stores, SERCA dysfunction, or reduced calcium influx through L-type channels. Each of these mechanisms disrupts the timely and synchronized release of calcium ions, postponing the interaction between contractile proteins and prolonging the time between electrical excitation and mechanical contraction. Understanding these processes is crucial for identifying therapeutic targets to mitigate latency and improve cardiac function in various pathological states.
Stretching Risks: Can It Cause Muscle Tears?
You may want to see also
Explore related products






![]()
Impaired Ion Channels: Dysfunctional ion channels disrupt electrical signaling, causing delayed depolarization and latency
Impaired ion channels play a critical role in the development of latency in cardiac muscle by disrupting the precise electrical signaling required for coordinated contractions. Cardiac muscle cells rely on the rapid and synchronized flow of ions, such as sodium (Na⁺), potassium (K�+), and calcium (Ca²⁺), to generate and propagate action potentials. These action potentials trigger muscle contraction. When ion channels malfunction, the normal flow of ions is hindered, leading to delayed depolarization—the process by which the cell’s membrane potential shifts from negative to positive, initiating contraction. Dysfunctional sodium channels, for instance, may fail to open or close properly, slowing the influx of Na⁺ ions during the initial phase of the action potential. This delay in depolarization disrupts the timing of electrical signals, causing latency in the contraction of cardiac muscle fibers.
Potassium channels are equally vital in repolarizing the cell membrane after depolarization, restoring it to its resting state and preparing it for the next cycle. Impaired potassium channels can prolong the repolarization phase, further contributing to latency. For example, mutations in potassium channel genes, such as those encoding Kv11.1 (hERG), can lead to long QT syndrome, a condition characterized by delayed repolarization and increased risk of arrhythmias. This prolongation of the action potential duration disrupts the synchronized electrical activity across the heart, resulting in inefficient and delayed contractions. The cumulative effect of such disruptions is a noticeable latency in cardiac muscle response, impairing overall heart function.
Calcium channels also play a pivotal role in cardiac muscle contraction, as calcium influx triggers the release of additional calcium from intracellular stores, initiating the mechanical contraction process. Dysfunctional calcium channels can impair this mechanism, leading to reduced or delayed calcium release. This not only slows the onset of contraction but also weakens the force of contraction, contributing to latency. Conditions like hypertrophic cardiomyopathy, where calcium channel function is compromised, exemplify how ion channel dysfunction directly translates to delayed and inefficient cardiac muscle performance.
Moreover, the interplay between different ion channels exacerbates the impact of individual channel impairments. For instance, a delay in sodium channel activation can cascade into delayed calcium channel opening, further prolonging the depolarization and contraction phases. This interconnectedness means that even a single dysfunctional ion channel can have widespread effects on electrical signaling and muscle contraction. Pharmacological interventions targeting these channels, such as calcium channel blockers or potassium channel modulators, are often used to mitigate latency, highlighting the central role of ion channels in maintaining cardiac rhythm.
In summary, impaired ion channels are a primary cause of latency in cardiac muscle due to their direct influence on electrical signaling and depolarization. Dysfunctional sodium, potassium, and calcium channels disrupt the timing and amplitude of action potentials, leading to delayed and inefficient contractions. Understanding these mechanisms is crucial for diagnosing and treating cardiac conditions associated with latency, emphasizing the importance of ion channel health in maintaining optimal heart function.
Muscle Cramps: ATP Deficiency or Something Else?
You may want to see also
Explore related products






![]()
Reduced ATP Levels: Low energy availability slows cross-bridge cycling, prolonging contraction and relaxation times
Reduced ATP levels in cardiac muscle cells play a critical role in causing latency by impairing the efficiency of cross-bridge cycling, a fundamental process in muscle contraction and relaxation. ATP (adenosine triphosphate) is the primary energy currency of cells, and its availability is essential for the proper functioning of the cardiac muscle. During contraction, myosin heads bind to actin filaments and pull them, a process fueled by ATP hydrolysis. When ATP levels are low, the rate at which myosin heads detach from actin and rebind for the next cycle is significantly slowed. This delay in cross-bridge cycling directly prolongs the duration of both the contraction (systole) and relaxation (diastole) phases of the cardiac cycle, leading to latency in cardiac muscle function.
The slowing of cross-bridge cycling due to reduced ATP levels has a cascading effect on cardiac muscle performance. Normally, ATP is rapidly regenerated through oxidative phosphorylation and glycolysis, ensuring continuous energy supply for muscle activity. However, in conditions such as ischemia, hypoxia, or metabolic disorders, ATP production is compromised. Without sufficient ATP, the myosin heads remain attached to actin longer than necessary, hindering the smooth transition between contraction and relaxation. This prolongation of mechanical events disrupts the synchronized beating of the heart, manifesting as latency in cardiac muscle response.
Another consequence of low ATP levels is the impaired function of calcium pumps, such as the sarcoplasmic reticulum (SR) ATPase, which are crucial for calcium homeostasis in cardiac cells. Calcium ions are essential for initiating contraction by binding to troponin and exposing myosin-binding sites on actin. After contraction, calcium is actively pumped back into the SR to allow relaxation. Reduced ATP availability slows this reuptake process, leading to elevated cytosolic calcium levels. Prolonged exposure to calcium delays relaxation and increases the time required for the muscle to prepare for the next contraction, further contributing to latency.
Furthermore, low ATP levels can activate stress pathways that exacerbate latency in cardiac muscle. Energy depletion triggers the accumulation of ADP and AMP, which activate AMP-activated protein kinase (AMPK). While AMPK helps restore energy balance by inhibiting ATP-consuming processes and enhancing ATP production, its activation can also lead to transient reductions in contractile function. This protective mechanism, though beneficial in the long term, can temporarily slow cross-bridge cycling and prolong contraction and relaxation times, adding to the latency observed in cardiac muscle under energy-deficient conditions.
In summary, reduced ATP levels directly and indirectly contribute to latency in cardiac muscle by slowing cross-bridge cycling and disrupting calcium handling. The energy-dependent nature of these processes means that even minor ATP deficits can have pronounced effects on cardiac function. Understanding this mechanism highlights the importance of maintaining adequate energy supply to the heart, particularly in clinical scenarios where ATP production is compromised. Addressing energy deficits through interventions such as improving oxygen delivery, metabolic support, or pharmacological therapies may help mitigate latency and restore optimal cardiac performance.
Nitrofurantoin's Musculoskeletal Pain: What You Need to Know
You may want to see also
Explore related products






![]()
Hypoxia Effects: Oxygen deprivation impairs metabolic processes, slowing muscle fiber response and increasing latency
Hypoxia, or oxygen deprivation, significantly impacts cardiac muscle function by disrupting essential metabolic processes. Cardiac muscle cells, like all cells, rely on oxygen to produce adenosine triphosphate (ATP) through oxidative phosphorylation. ATP is the primary energy currency required for muscle contraction and relaxation. When oxygen levels decrease, as in hypoxic conditions, the efficiency of oxidative phosphorylation declines. This forces cardiac muscle cells to shift to anaerobic glycolysis, a less efficient energy production pathway. Anaerobic glycolysis not only yields significantly less ATP but also produces lactic acid as a byproduct, which can further impair cellular function. This energy deficit directly slows the muscle fibers' ability to respond to electrical signals, thereby increasing latency in cardiac muscle contraction.
The reduced ATP availability under hypoxic conditions compromises the function of critical proteins involved in muscle contraction, such as actin and myosin. These proteins require ATP to detach and reattach during the contraction cycle. With insufficient ATP, the cross-bridge cycling between actin and myosin slows down, leading to delayed muscle fiber shortening. Additionally, calcium handling, a process vital for initiating and terminating cardiac muscle contractions, is ATP-dependent. Hypoxia impairs the activity of the sarcoplasmic reticulum (SR) ATPase, which is responsible for pumping calcium back into the SR after contraction. This results in prolonged calcium transient durations, further slowing relaxation and increasing latency in cardiac muscle response.
Hypoxia also triggers intracellular signaling pathways that exacerbate latency in cardiac muscle. One such pathway involves the activation of hypoxia-inducible factors (HIFs), which regulate gene expression in response to low oxygen levels. While HIFs can upregulate genes involved in angiogenesis and glycolysis, they may also downregulate genes essential for maintaining optimal cardiac function. For instance, HIF activation can lead to the suppression of genes encoding for contractile proteins or ion channels, indirectly contributing to slower muscle fiber response. Furthermore, hypoxia-induced oxidative stress damages cellular components, including proteins and membranes, impairing their function and adding to the overall latency in cardiac muscle contraction.
Another critical effect of hypoxia is its impact on electrophysiological properties of cardiac muscle cells. Oxygen deprivation alters the function of ion channels and transporters, which are crucial for generating and propagating action potentials. For example, hypoxia can reduce the activity of sodium-potassium ATPase pumps, leading to intracellular sodium and calcium overload. This disrupts the resting membrane potential and slows the repolarization phase of the action potential, delaying the next contraction cycle. The cumulative effect of these electrophysiological changes is an increase in latency, as the cardiac muscle fibers take longer to respond to electrical stimuli.
In summary, hypoxia-induced oxygen deprivation profoundly impairs metabolic processes in cardiac muscle, leading to a cascade of events that increase latency. From reduced ATP production and compromised protein function to altered gene expression and electrophysiological disturbances, hypoxia affects multiple levels of cardiac muscle physiology. Understanding these mechanisms is crucial for developing strategies to mitigate the effects of hypoxia on cardiac function, particularly in clinical scenarios such as ischemia or high-altitude exposure. Addressing hypoxia-related latency in cardiac muscle is essential for maintaining cardiovascular health and preventing complications associated with oxygen deprivation.
Health Anxiety: Can It Weaken Your Muscles?
You may want to see also
Explore related products






![]()
Aging Impact: Age-related fibrosis and stiffening reduce cardiac muscle elasticity, delaying contraction and relaxation
As we age, the heart undergoes structural and functional changes that contribute to latency in cardiac muscle performance. One of the primary aging impacts is the development of age-related fibrosis, where excessive collagen deposition occurs in the extracellular matrix of the myocardium. This fibrotic tissue replaces functional cardiomyocytes, leading to a loss of contractile units. Since collagen is inherently stiffer than muscle tissue, this substitution reduces the overall elasticity of the cardiac muscle. Elasticity is crucial for efficient contraction and relaxation, as it allows the heart to recoil and refill with blood during diastole. With diminished elasticity, the heart’s ability to deform and return to its original shape is compromised, resulting in delayed relaxation and slower ventricular filling.
In addition to fibrosis, age-related stiffening of the cardiac muscle further exacerbates latency. This stiffening is partly due to the cross-linking of collagen fibers and the accumulation of advanced glycation end products (AGEs), which increase tissue rigidity. Stiff myocardium requires more time and energy to stretch and contract, leading to prolonged contraction and relaxation phases. The delayed contraction reduces cardiac output, while the delayed relaxation impairs diastolic function, causing blood to back up in the pulmonary and systemic circulation. This combination of reduced elasticity and increased stiffness disrupts the synchronized electrical and mechanical activity of the heart, contributing to latency in cardiac muscle response.
The reduction in cardiac muscle elasticity due to aging also impairs the Frank-Starling mechanism, a critical process that ensures the heart pumps an appropriate volume of blood in response to venous return. Normally, increased stretching of cardiomyocytes during diastole enhances their contractile force, maintaining cardiac output. However, age-related fibrosis and stiffening limit the heart’s ability to stretch adequately, weakening the Frank-Starling response. This impairment results in decreased stroke volume and overall cardiac efficiency, further delaying contraction and relaxation. As a consequence, the heart struggles to meet the body’s demands, particularly during physical exertion or stress.
Moreover, the microcirculatory changes associated with aging compound the effects of fibrosis and stiffening. Reduced capillary density and impaired endothelial function in aged hearts limit oxygen and nutrient delivery to cardiomyocytes, hindering their ability to contract and relax efficiently. This ischemic environment contributes to cellular dysfunction and apoptosis, accelerating the loss of contractile tissue. The cumulative effect is a slower, less coordinated cardiac cycle, with latency manifesting as prolonged QRS complexes and PR intervals on electrocardiograms. These electrophysiological delays reflect the underlying mechanical inefficiencies caused by age-related fibrosis and stiffening.
Finally, the neurohormonal adaptations to aging further contribute to latency in cardiac muscle. As the heart struggles with reduced elasticity and stiffening, compensatory mechanisms such as increased sympathetic nervous system activity and renin-angiotensin-aldosterone system (RAAS) activation are triggered. While these mechanisms aim to maintain blood pressure and cardiac output, they also impose additional stress on the myocardium, promoting further fibrosis and hypertrophy. This vicious cycle exacerbates stiffness and reduces elasticity, prolonging contraction and relaxation times. Thus, age-related fibrosis and stiffening are central to the latency observed in cardiac muscle, with far-reaching implications for cardiovascular health and function in older adults.
Adrenal Fatigue: Can It Cause Facial Muscle Loss?
You may want to see also
Frequently asked questions
Latency in cardiac muscle refers to a delay in the electrical or mechanical response of the heart tissue. It can affect heart function by disrupting the synchronized contraction of the heart chambers, leading to arrhythmias, reduced cardiac output, or inefficient pumping of blood.
Primary causes include ischemia (reduced blood flow), electrolyte imbalances (e.g., low potassium or calcium), myocardial scarring from previous heart attacks, and certain medications that alter electrical conduction in the heart.
Ischemia reduces oxygen and nutrient supply to cardiac muscle cells, impairing their ability to generate and propagate electrical signals. This leads to delayed or blocked conduction, resulting in latency and potential arrhythmias.
Yes, aging can cause latency due to fibrosis (scarring) and stiffening of cardiac tissue, which slows electrical conduction. Additionally, age-related changes in ion channel function and reduced cellular metabolism contribute to delayed responses in cardiac muscle.