Smooth muscle cell relaxation is a critical physiological process regulated by various signaling pathways and molecular mechanisms. Among the events that directly result in smooth muscle cell relaxation, the activation of cyclic nucleotide-dependent pathways, particularly the increase in cyclic guanosine monophosphate (cGMP) levels, plays a pivotal role. The binding of nitric oxide (NO) to the enzyme soluble guanylate cyclase (sGC) stimulates cGMP production, which in turn activates protein kinase G (PKG). PKG subsequently phosphorylates target proteins, leading to a reduction in intracellular calcium concentration and the dephosphorylation of myosin light chains, ultimately causing smooth muscle relaxation. This mechanism is central to processes such as vasodilation, bronchial dilation, and gastrointestinal motility, highlighting its significance in maintaining homeostasis and responding to physiological stimuli.
Explore related products






What You'll Learn
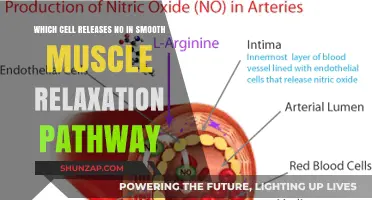
- Nitric Oxide Release: Endothelial cells release nitric oxide, activating guanylate cyclase in smooth muscle cells
- cGMP Increase: Elevated cGMP levels activate protein kinase G, reducing calcium ion concentration
- Calcium Channel Blockade: Voltage-gated calcium channels are blocked, decreasing intracellular calcium levels
- Beta-2 Agonists: Stimulation of beta-2 adrenergic receptors activates adenylate cyclase, increasing cAMP levels
- Prostaglandin E1 Action: Binds to IP receptors, raising cAMP, leading to decreased calcium sensitivity
![]()
Nitric Oxide Release: Endothelial cells release nitric oxide, activating guanylate cyclase in smooth muscle cells
Nitric oxide (NO) is a pivotal signaling molecule in vascular biology, playing a central role in regulating smooth muscle cell relaxation. Endothelial cells lining blood vessels synthesize NO in response to various stimuli, such as shear stress from blood flow or chemical signals like acetylcholine. Once released, NO diffuses rapidly into adjacent smooth muscle cells, where it binds to the enzyme soluble guanylate cyclase (sGC). This binding triggers a cascade of events that culminates in smooth muscle relaxation, a process essential for vasodilation and maintaining healthy blood flow.
The mechanism by which NO induces relaxation is both elegant and efficient. Upon activation, sGC catalyzes the conversion of guanosine triphosphate (GTP) to cyclic guanosine monophosphate (cGMP). Elevated cGMP levels act as a second messenger, activating protein kinase G (PKG), which phosphorylates target proteins. One critical target is the calcium-sensitive protein calmodulin, which, when phosphorylated, reduces intracellular calcium levels. Lower calcium concentrations lead to decreased activation of myosin light-chain kinase, a key enzyme in smooth muscle contraction. As a result, the smooth muscle cells relax, allowing blood vessels to dilate.
Clinically, understanding this pathway has profound implications. For instance, drugs like nitroglycerin, used to treat angina, work by releasing NO or its metabolites, which then activate sGC in smooth muscle cells. Similarly, phosphodiesterase type 5 (PDE5) inhibitors, such as sildenafil, enhance the effects of NO by preventing cGMP breakdown, thereby prolonging smooth muscle relaxation. However, excessive NO production or dysregulation of this pathway can lead to hypotension or other vascular complications, underscoring the need for precise therapeutic targeting.
Practical considerations for optimizing NO-mediated smooth muscle relaxation include lifestyle factors. Regular physical activity increases shear stress on endothelial cells, promoting NO release. Dietary nitrates, found in foods like beets and leafy greens, serve as precursors for NO synthesis and can enhance vascular function. Conversely, smoking and hypertension impair endothelial NO production, highlighting the importance of managing these risk factors. For older adults or individuals with cardiovascular disease, combining pharmacological interventions with lifestyle modifications can maximize the benefits of this pathway.
In summary, the release of NO by endothelial cells and its subsequent activation of guanylate cyclase in smooth muscle cells represent a fundamental mechanism of vascular relaxation. This process is not only critical for physiological blood flow regulation but also a target for therapeutic interventions in cardiovascular disease. By understanding and supporting this pathway, clinicians and individuals alike can promote vascular health and mitigate the risks associated with impaired smooth muscle relaxation.
Effective Techniques to Relax and Soothe Your Back Muscles
You may want to see also
Explore related products






![]()
cGMP Increase: Elevated cGMP levels activate protein kinase G, reducing calcium ion concentration
Elevated levels of cyclic guanosine monophosphate (cGMP) play a pivotal role in the relaxation of smooth muscle cells, a process critical in various physiological functions such as vasodilation and bronchodilation. When cGMP levels increase, it directly activates protein kinase G (PKG), an enzyme that initiates a cascade of events leading to smooth muscle relaxation. This mechanism is particularly significant in vascular smooth muscle, where it helps regulate blood flow and pressure. For instance, in the context of erectile dysfunction treatments, drugs like sildenafil (Viagra) enhance cGMP signaling by inhibiting its breakdown, thereby promoting sustained smooth muscle relaxation in the corpus cavernosum.
The activation of PKG by cGMP triggers a series of biochemical reactions that ultimately reduce intracellular calcium ion concentration. Calcium ions are essential for smooth muscle contraction, as they bind to calmodulin and activate myosin light-chain kinase (MLCK), leading to cross-bridge cycling and muscle fiber shortening. By lowering calcium levels, PKG effectively counteracts this process. Specifically, PKG phosphorylates and activates phosphodiesterases, which degrade cyclic adenosine monophosphate (cAMP), and inhibits calcium channels, reducing calcium influx. Additionally, PKG promotes the sequestration of calcium ions into the sarcoplasmic reticulum via phosphorylation of phospholamban, further diminishing cytosolic calcium availability.
From a practical standpoint, understanding this pathway has significant implications for therapeutic interventions. For example, in patients with pulmonary arterial hypertension, medications like riociguat directly stimulate soluble guanylate cyclase to increase cGMP production, thereby enhancing PKG activity and reducing vascular resistance. Dosage adjustments for such drugs are critical, as excessive cGMP elevation can lead to hypotension or syncope. Clinicians often start with lower doses (e.g., 1.5 mg tid for riociguat) and titrate upward based on patient response and tolerability, particularly in older adults or those with comorbidities.
Comparatively, this cGMP-PKG pathway contrasts with other relaxation mechanisms, such as those mediated by nitric oxide (NO) or beta-adrenergic signaling. While NO also increases cGMP via guanylate cyclase activation, the PKG-mediated reduction in calcium ions is a more direct and localized mechanism. In contrast, beta-adrenergic signaling primarily acts through cAMP and protein kinase A (PKA), which phosphorylates MLCK to inhibit contraction. The cGMP pathway’s specificity makes it a targeted therapeutic option, particularly in conditions where calcium overload or hypercontractility is a primary issue, such as in asthma or hypertension.
In conclusion, the increase in cGMP levels and subsequent activation of PKG represent a critical event directly resulting in smooth muscle cell relaxation. By reducing intracellular calcium ion concentration through multiple mechanisms, this pathway effectively counteracts the contractile machinery of smooth muscle. Its therapeutic exploitation in conditions ranging from erectile dysfunction to pulmonary hypertension underscores its physiological and clinical importance. For practitioners, understanding this pathway enables precise pharmacological interventions, emphasizing the need for tailored dosing and patient monitoring to optimize outcomes while minimizing adverse effects.
Is Tylenol Extra Strength a Muscle Relaxer? Facts Revealed
You may want to see also
Explore related products






![]()
Calcium Channel Blockade: Voltage-gated calcium channels are blocked, decreasing intracellular calcium levels
Smooth muscle cell relaxation is a critical process in various physiological functions, from blood pressure regulation to gastrointestinal motility. One direct mechanism that triggers this relaxation is the blockade of voltage-gated calcium channels, which reduces intracellular calcium levels. This process is central to understanding how smooth muscle cells transition from a contracted to a relaxed state. By inhibiting calcium influx, the cell’s ability to maintain contraction is compromised, leading to relaxation. This mechanism is not only a fundamental biological process but also a therapeutic target in treating conditions like hypertension and angina.
Analytically, voltage-gated calcium channels play a pivotal role in smooth muscle contraction by allowing calcium ions to enter the cell, which then bind to calmodulin and activate myosin light-chain kinase. This enzyme phosphorylates myosin, enabling actin-myosin cross-bridge formation and muscle contraction. When these channels are blocked—either pharmacologically or through physiological mechanisms—calcium entry is reduced, and the intracellular calcium concentration drops. Without sufficient calcium, the contraction machinery cannot function, and relaxation ensues. For example, calcium channel blockers like nifedipine (a dihydropyridine) are commonly prescribed to treat hypertension by relaxing arterial smooth muscle, thereby reducing vascular resistance and lowering blood pressure.
Instructively, calcium channel blockade is achieved through specific drugs that target L-type voltage-gated calcium channels, which are predominant in vascular and cardiac smooth muscle. These drugs, such as verapamil and diltiazem, bind to the channels and prevent their opening, even in the presence of membrane depolarization. Dosage varies by condition and patient age: for adults with hypertension, nifedipine is often initiated at 30–60 mg/day, while elderly patients may require lower doses due to reduced metabolic clearance. It’s crucial to monitor for side effects like dizziness and edema, as these drugs can cause systemic vasodilation. Practical tips include taking the medication with food to enhance absorption and avoiding grapefruit juice, which can inhibit drug metabolism.
Comparatively, calcium channel blockade stands apart from other smooth muscle relaxation mechanisms, such as nitric oxide (NO) signaling or beta-adrenergic receptor activation. While NO increases cyclic GMP levels to reduce calcium sensitivity, and beta-agonists activate protein kinase A to inhibit calcium release, calcium channel blockers directly limit calcium entry. This makes them particularly effective in conditions where calcium overload is the primary issue, such as in coronary artery spasm. However, their systemic effects—like reflex tachycardia due to reduced blood pressure—highlight the need for careful patient selection and monitoring.
Descriptively, the process of calcium channel blockade is akin to silencing a critical signal in the smooth muscle cell’s contraction cascade. Imagine a well-orchestrated symphony where calcium ions are the conductors, guiding the contraction machinery. When the channels are blocked, the conductors are muted, and the orchestra—the actin and myosin filaments—gradually cease their activity. This metaphor underscores the elegance and precision of this mechanism, which has been harnessed in medicine to alleviate suffering and improve quality of life for millions of patients worldwide. Understanding this process not only deepens our appreciation of cellular biology but also empowers clinicians to use these drugs effectively and safely.
Muscle Relaxants: Understanding the Mandatory Cautionary Label on Prescriptions
You may want to see also
Explore related products



$9.99



![]()
Beta-2 Agonists: Stimulation of beta-2 adrenergic receptors activates adenylate cyclase, increasing cAMP levels
Beta-2 agonists are a class of drugs primarily used to treat respiratory conditions such as asthma and chronic obstructive pulmonary disease (COPD). Their mechanism of action is both precise and fascinating, directly leading to smooth muscle cell relaxation in the airways. When a beta-2 agonist, like albuterol or salmeterol, binds to beta-2 adrenergic receptors on the surface of smooth muscle cells, it triggers a cascade of intracellular events. The first critical step is the activation of adenylate cyclase, an enzyme that converts ATP to cyclic adenosine monophosphate (cAMP). This increase in cAMP levels acts as a secondary messenger, initiating a series of reactions that ultimately lead to muscle relaxation. For instance, cAMP activates protein kinase A (PKA), which phosphorylates proteins involved in calcium regulation, reducing intracellular calcium levels and causing the smooth muscle to relax.
Analyzing the clinical application of beta-2 agonists reveals their effectiveness in rapidly relieving bronchoconstriction. Short-acting beta-2 agonists (SABAs), such as albuterol, are commonly used as rescue medications for acute symptoms, with a typical inhaled dose ranging from 90 to 108 mcg for adults. Long-acting beta-2 agonists (LABAs), like salmeterol or formoterol, are used for maintenance therapy in patients with persistent symptoms, often in combination with inhaled corticosteroids. It’s crucial to note that while LABAs provide sustained bronchodilation, they should not be used as monotherapy due to the risk of severe asthma exacerbations. This distinction highlights the importance of tailoring treatment to the patient’s specific needs and disease severity.
From a comparative perspective, beta-2 agonists stand out among bronchodilators due to their direct and rapid action on smooth muscle cells. Unlike anticholinergics, which block muscarinic receptors to inhibit bronchoconstriction, beta-2 agonists actively stimulate relaxation by increasing cAMP levels. This makes them particularly effective for quick relief during acute episodes. However, their mechanism also underscores the need for careful dosing, as excessive stimulation of beta-2 receptors can lead to side effects such as tremors, palpitations, and hypokalemia. For children, dosages are adjusted based on age and weight, with pediatric inhalers often delivering lower doses (e.g., 45–90 mcg per puff) to minimize adverse effects.
A persuasive argument for the use of beta-2 agonists lies in their ability to improve quality of life for patients with respiratory conditions. By directly targeting the biochemical pathway that leads to smooth muscle relaxation, these drugs provide immediate and measurable relief. For example, studies have shown that albuterol can increase forced expiratory volume in one second (FEV1) by 15–20% within minutes of administration. This rapid onset of action is particularly valuable during asthma attacks, where timely intervention can prevent hospitalization. However, patients must be educated on proper inhaler technique and the importance of adhering to prescribed regimens to maximize benefits while minimizing risks.
In conclusion, the stimulation of beta-2 adrenergic receptors by agonists, leading to increased cAMP levels, is a direct and efficient pathway to smooth muscle cell relaxation. This mechanism not only explains their efficacy in treating respiratory conditions but also guides their appropriate use in clinical practice. Whether as a rescue medication or part of long-term management, beta-2 agonists remain a cornerstone of therapy for millions of patients worldwide. Understanding their action at the molecular level empowers healthcare providers to optimize treatment outcomes while ensuring patient safety.
Is Xanax a Skeletal Muscle Relaxant? Unraveling the Facts
You may want to see also
Explore related products






![]()
Prostaglandin E1 Action: Binds to IP receptors, raising cAMP, leading to decreased calcium sensitivity
Prostaglandin E1 (PGE1) is a potent vasodilator and smooth muscle relaxant, playing a critical role in regulating vascular tone and tissue perfusion. Its mechanism of action hinges on a precise molecular cascade: binding to IP receptors, activating adenylate cyclase, and subsequently elevating intracellular cyclic adenosine monophosphate (cAMP) levels. This surge in cAMP initiates a series of events that culminate in reduced calcium sensitivity within smooth muscle cells, directly promoting relaxation. Understanding this pathway is essential for clinicians and researchers, particularly in contexts like erectile dysfunction treatment, where PGE1 is administered via intracavernosal injection at doses ranging from 2.5 to 40 μg, depending on patient response and tolerance.
The process begins with PGE1’s interaction with IP receptors, G protein-coupled receptors located on the cell membrane of smooth muscle cells. Upon binding, the receptor activates Gs proteins, which stimulate adenylate cyclase to convert ATP into cAMP. Elevated cAMP levels then activate protein kinase A (PKA), a key enzyme that phosphorylates target proteins, including phospholamban and myosin light chain kinase (MLCK). Phosphorylation of phospholamban enhances calcium uptake into the sarcoplasmic reticulum, while phosphorylation of MLCK reduces its activity. These actions collectively decrease cytosolic calcium availability, impairing the calcium-calmodulin complex’s ability to activate myosin light chain phosphatase and initiate muscle contraction. The result is smooth muscle relaxation, a principle exploited in therapeutic applications like the treatment of peripheral arterial disease or pulmonary hypertension.
Clinically, PGE1’s ability to modulate calcium sensitivity offers a targeted approach to managing conditions characterized by excessive smooth muscle tone. For instance, in patients with Raynaud’s phenomenon, intravenous PGE1 at doses of 0.5 to 2 ng/kg/min improves digital perfusion by relaxing arterial smooth muscle. However, practitioners must balance efficacy with side effects, such as flushing, headache, and hypotension, which are dose-dependent and more common at higher concentrations. Monitoring hemodynamic parameters during infusion is critical, particularly in elderly patients or those with cardiovascular comorbidities, where the risk of adverse events may be elevated.
Comparatively, PGE1’s mechanism contrasts with other smooth muscle relaxants like nitrates, which act by releasing nitric oxide to activate soluble guanylate cyclase and increase cGMP. While both pathways reduce calcium sensitivity, PGE1’s reliance on cAMP provides a distinct pharmacological profile, making it a valuable alternative in nitrate-refractory cases. For example, in patients with coronary artery disease, PGE1’s direct action on IP receptors can offer vasodilatory benefits without cross-tolerance issues associated with chronic nitrate use. This comparative advantage underscores the importance of understanding PGE1’s unique mechanism in tailoring therapeutic strategies.
In practical terms, optimizing PGE1’s efficacy requires careful consideration of dosage, route of administration, and patient-specific factors. Intracavernosal injections for erectile dysfunction should start at 5 μg and titrated upward in 2.5 μg increments until a rigid erection is achieved or side effects become limiting. For systemic use, such as in neonatal ductus arteriosus patency, PGE1 is administered intravenously at 0.05 to 0.1 μg/kg/min, with close monitoring of oxygenation and perfusion parameters. Adjunctive therapies, such as concurrent use of phosphodiesterase inhibitors to prolong cAMP activity, may enhance outcomes but require cautious dosing to avoid synergistic side effects. By mastering these nuances, clinicians can harness PGE1’s calcium-desensitizing action to achieve precise and effective smooth muscle relaxation in diverse clinical scenarios.
Effective Ways to Eliminate Dark Circles with Muscle Relaxers
You may want to see also
Frequently asked questions
Nitric oxide (NO) is a key signaling molecule that directly results in smooth muscle cell relaxation by diffusing into the muscle cells and activating soluble guanylate cyclase (sGC). This enzyme increases cyclic guanosine monophosphate (cGMP) levels, which activates protein kinase G (PKG). PKG then phosphorylates target proteins, leading to decreased calcium levels and relaxation of the smooth muscle.
Activation of beta-2 adrenergic receptors by catecholamines like epinephrine or drugs like salbutamol stimulates adenylate cyclase, increasing cyclic adenosine monophosphate (cAMP) levels. cAMP activates protein kinase A (PKA), which phosphorylates proteins involved in calcium regulation. This reduces intracellular calcium, leading to smooth muscle cell relaxation.
Prostacyclin (PGI2) binds to IP receptors on smooth muscle cells, activating adenylate cyclase and increasing cAMP levels. Elevated cAMP activates PKA, which phosphorylates proteins that reduce intracellular calcium concentration. This decrease in calcium results in the dephosphorylation of myosin light chains, leading to smooth muscle cell relaxation.