Myasthenia gravis is an autoimmune disorder that leads to muscle weakness by disrupting the communication between nerves and muscles. In this condition, the immune system mistakenly produces antibodies that attack the acetylcholine receptors—essential proteins on muscle cells that receive signals from nerve endings. This interference reduces the ability of muscles to contract effectively, resulting in weakness that typically worsens with activity and improves with rest. Commonly affected muscles include those controlling eye and facial movements, swallowing, and breathing, though any voluntary muscle can be impacted. The progressive nature of this weakness, particularly during prolonged use, is a hallmark of the disease and underscores the critical role of acetylcholine receptors in neuromuscular transmission.
| Characteristics | Values |
|---|---|
| Autoimmune Attack | Myasthenia Gravis (MG) is an autoimmune disease where the immune system mistakenly attacks healthy tissues. In MG, antibodies target the acetylcholine receptors (AChR) at the neuromuscular junction. |
| Acetylcholine Receptor Blockade | Antibodies bind to AChR, blocking the binding of acetylcholine (ACh), a neurotransmitter essential for muscle contraction. This prevents the transmission of nerve impulses to muscles. |
| Complement-Mediated Damage | Antibody binding can activate the complement system, leading to damage and destruction of the neuromuscular junction, further reducing muscle activation. |
| Reduced Acetylcholine Release | In some cases, MG may involve antibodies against muscle-specific kinase (MuSK) or low-density lipoprotein receptor-related protein 4 (LRP4), which are crucial for AChR clustering and function, indirectly reducing ACh release. |
| Fatigable Weakness | Muscle weakness in MG is typically worse with repeated use and improves with rest, as the neuromuscular junction becomes less effective with prolonged activity. |
| Affected Muscles | MG primarily affects voluntary muscles, particularly those controlling eye and eyelid movement, facial expression, chewing, talking, and swallowing. Limb muscles are often involved but usually later in the disease course. |
| Thymic Abnormalities | Many MG patients have thymic abnormalities, such as hyperplasia or tumors, which may contribute to the production of autoantibodies against AChR. |
| Fluctuating Symptoms | Symptoms can vary in severity throughout the day, often worsening in the evening or after physical activity. |
| Response to Anticholinesterases | Muscle weakness in MG often responds to anticholinesterase medications, which increase ACh levels at the neuromuscular junction, temporarily improving muscle strength. |
| Myasthenic Crisis | Severe muscle weakness can lead to a life-threatening condition called myasthenic crisis, where respiratory muscles are affected, requiring immediate medical intervention. |
Explore related products






What You'll Learn
![]()
Autoimmune Attack on Acetylcholine Receptors
Myasthenia gravis (MG) is a chronic autoimmune disorder characterized by muscle weakness and fatigue, primarily due to an autoimmune attack on acetylcholine receptors (AChRs) at the neuromuscular junction (NMJ). In a healthy individual, nerve signals are transmitted to muscles via the release of acetylcholine (ACh), a neurotransmitter that binds to AChRs on muscle fibers, triggering contraction. However, in MG, the immune system mistakenly produces antibodies that target and disrupt these AChRs, impairing muscle activation.
The autoimmune attack begins when the body's immune system incorrectly identifies AChRs as foreign invaders. This leads to the production of autoantibodies, primarily IgG antibodies, which bind to the AChRs on the postsynaptic membrane of muscle fibers. This binding has multiple detrimental effects: it blocks ACh from attaching to the receptors, reduces the number of functional receptors by accelerating their degradation, and triggers complement-mediated damage to the postsynaptic membrane. As a result, the efficiency of signal transmission from nerve to muscle is severely compromised, leading to muscle weakness.
The reduction in functional AChRs means that even when ACh is released, it cannot effectively stimulate muscle contraction. Over time, the muscle fibers become less responsive to neural input, causing the hallmark symptoms of MG, such as drooping eyelids, difficulty swallowing, and generalized muscle fatigue. The severity of muscle weakness correlates with the extent of AChR destruction and the body's ability to compensate for the loss of functional receptors.
Interestingly, not all patients with MG have antibodies targeting AChRs. A smaller subset has antibodies against muscle-specific kinase (MuSK), a protein essential for clustering AChRs at the NMJ. However, the majority of MG cases (approximately 85%) are directly linked to anti-AChR antibodies, underscoring the central role of this autoimmune attack in the pathogenesis of the disease.
Treatment strategies for MG often focus on mitigating the effects of this autoimmune attack. Medications such as acetylcholinesterase inhibitors (e.g., pyridostigmine) enhance ACh availability at the NMJ, temporarily improving muscle strength. Immunosuppressive therapies, including corticosteroids and biologics, aim to reduce the production of autoantibodies and slow the destruction of AChRs. In severe cases, plasmapheresis or intravenous immunoglobulin (IVIG) may be used to remove or neutralize circulating autoantibodies, providing rapid relief from symptoms. Understanding the autoimmune attack on AChRs is thus crucial for both diagnosing MG and developing effective treatment approaches.
Nerve-Muscle Connection: Why Twitching Occurs
You may want to see also
Explore related products


$9.99




![]()
Impaired Neuromuscular Junction Signaling
Myasthenia gravis (MG) is an autoimmune disorder characterized by muscle weakness and fatigue, primarily due to impaired neuromuscular junction (NMJ) signaling. The NMJ is the critical interface where motor neurons communicate with skeletal muscles, enabling movement. In MG, this communication is disrupted, leading to inefficient muscle activation. The root cause lies in the autoimmune attack on key components of the NMJ, particularly the acetylcholine receptors (AChRs) and, in some cases, muscle-specific kinase (MuSK) proteins. These attacks result in a reduction in the number of functional AChRs, which are essential for transmitting signals from the nerve to the muscle.
The autoimmune response in MG is driven by autoantibodies targeting AChRs or MuSK. When these autoantibodies bind to AChRs, they trigger a cascade of events that impair NMJ function. One mechanism is antibody-mediated internalization and degradation of AChRs, reducing their availability on the muscle membrane. Additionally, autoantibodies can activate the complement system, leading to direct damage and destruction of the postsynaptic membrane. This loss of AChRs diminishes the muscle’s ability to detect and respond to acetylcholine (ACh), the neurotransmitter released by motor neurons. As a result, the signal from the nerve to the muscle is weakened, leading to suboptimal muscle contraction.
Another critical aspect of impaired NMJ signaling in MG is the reduced release of acetylcholine from motor neuron terminals. While less common, some patients with MG have autoantibodies targeting MuSK, a protein essential for maintaining the structure and function of the NMJ. MuSK is involved in clustering AChRs and ensuring proper synaptic organization. When MuSK is compromised, AChR clustering is disrupted, further impairing signal transmission. This disruption exacerbates the weakness in muscle activation, as the NMJ fails to maintain its integrity and functionality.
The cumulative effect of these autoimmune attacks is a decrease in the safety factor of neuromuscular transmission. Normally, the NMJ ensures reliable muscle activation by releasing more ACh than is minimally required to trigger a muscle fiber contraction. In MG, the reduced number of functional AChRs and impaired NMJ function lower this safety factor, making muscle activation less reliable, especially during repeated or sustained activity. This is why MG patients experience muscle weakness that worsens with use and improves with rest.
In summary, impaired NMJ signaling in MG is primarily driven by autoimmune-mediated damage to AChRs and, in some cases, MuSK proteins. This damage reduces the number of functional AChRs, disrupts synaptic organization, and lowers the safety factor of neuromuscular transmission. The result is inefficient signal transmission from the nerve to the muscle, leading to the characteristic muscle weakness and fatigue observed in MG. Understanding these mechanisms is crucial for developing targeted therapies to restore NMJ function and alleviate symptoms in affected individuals.
Heat-Induced Muscle Cramps: Causes and Prevention Strategies Explained
You may want to see also
Explore related products






![]()
Reduced Muscle Fiber Stimulation
Myasthenia gravis (MG) is an autoimmune disorder that primarily affects the neuromuscular junction, the critical site where nerve cells communicate with muscle fibers to initiate movement. Central to the muscle weakness characteristic of MG is the concept of reduced muscle fiber stimulation. This occurs due to the disruption of normal signaling between nerves and muscles, leading to inadequate activation of muscle fibers. Below is a detailed exploration of this mechanism.
In a healthy individual, muscle contraction begins when a nerve impulse travels to the neuromuscular junction, releasing a neurotransmitter called acetylcholine (ACh). ACh binds to receptors on the muscle fiber, known as nicotinic acetylcholine receptors (AChRs), triggering a series of events that result in muscle contraction. In myasthenia gravis, however, the immune system mistakenly produces antibodies that target and attack these AChRs. This autoimmune response leads to the destruction or dysfunction of the receptors, reducing their availability for ACh binding. As a result, the signal from the nerve to the muscle is significantly weakened, leading to reduced muscle fiber stimulation.
The reduction in AChR function is further exacerbated by other immune-related mechanisms. For instance, activating complement proteins—part of the immune system—can cause damage to the postsynaptic membrane, where AChRs are located. Additionally, antibodies may accelerate the degradation of ACh or block its release, further diminishing the neurotransmitter’s ability to activate the remaining receptors. These processes collectively impair the efficiency of neuromuscular transmission, ensuring that even when ACh is released, it cannot adequately stimulate muscle fibers.
Another factor contributing to reduced muscle fiber stimulation in MG is the phenomenon of receptor desensitization. Even if some AChRs remain functional, their repeated exposure to ACh in the presence of antibodies can lead to a state of desensitization, where the receptors become less responsive to the neurotransmitter. This desensitization reduces the likelihood of muscle fiber activation, even when ACh is present in sufficient quantities. Over time, the cumulative effect of receptor loss, damage, and desensitization results in a profound decrease in muscle fiber stimulation, manifesting as muscle weakness and fatigue.
The impact of reduced muscle fiber stimulation is particularly noticeable in muscles that are frequently used or require sustained contraction, such as those involved in eye movement, facial expression, swallowing, and limb movement. These muscles are more susceptible to fatigue because the demand for repeated stimulation exceeds the compromised capacity of the neuromuscular junction to transmit signals effectively. As a result, patients with MG often experience fluctuating weakness that worsens with activity and improves with rest, reflecting the transient nature of muscle fiber stimulation in this condition.
In summary, reduced muscle fiber stimulation in myasthenia gravis arises from the autoimmune-mediated destruction, dysfunction, and desensitization of AChRs at the neuromuscular junction. This impairment disrupts the normal signaling process between nerves and muscles, leading to inadequate activation of muscle fibers and the characteristic muscle weakness of the disease. Understanding this mechanism is crucial for developing targeted therapies that aim to restore neuromuscular transmission and alleviate symptoms in affected individuals.
Understanding Weak Muscle Contractions: Causes and Contributing Factors
You may want to see also
Explore related products






![]()
Antibody-Induced Receptor Destruction
Myasthenia gravis (MG) is an autoimmune disorder characterized by muscle weakness and fatigue, primarily caused by the disruption of communication between nerve cells and muscles. A key mechanism underlying this disruption is antibody-induced receptor destruction, which plays a central role in the pathogenesis of the disease. In MG, the immune system mistakenly produces antibodies that target and attack critical components of the neuromuscular junction (NMJ), the site where nerve cells communicate with muscle fibers. Specifically, these autoantibodies target the nicotinic acetylcholine receptors (AChRs) located on the muscle cell membrane. AChRs are essential for transmitting signals from nerves to muscles, enabling muscle contraction. When these receptors are destroyed or impaired, the muscle’s ability to respond to nerve signals is severely compromised, leading to weakness and fatigue.
The process of antibody-induced receptor destruction begins with the production of autoantibodies, primarily IgG antibodies, by aberrant B cells. These antibodies bind to the extracellular domain of AChRs with high specificity. Once bound, the antibodies initiate a cascade of events that lead to the degradation or internalization of the receptors. One mechanism involves the activation of the complement system, a part of the immune system that can form membrane attack complexes, causing direct damage to the muscle cell membrane and the destruction of AChRs. Additionally, antibody binding can trigger endocytosis, where the receptor-antibody complex is internalized and degraded within the muscle cell, further reducing the number of functional AChRs available for signal transmission.
Another critical aspect of antibody-induced receptor destruction is the modulation of receptor function. Even if the receptors are not completely destroyed, the binding of autoantibodies can interfere with their ability to open ion channels in response to acetylcholine, the neurotransmitter released by nerve cells. This interference disrupts the normal flow of ions across the muscle cell membrane, preventing the generation of an action potential and subsequent muscle contraction. Over time, the cumulative effect of receptor destruction and functional impairment leads to a significant reduction in the muscle’s ability to contract efficiently, manifesting as the characteristic muscle weakness seen in MG.
The impact of antibody-induced receptor destruction is particularly pronounced in muscles that are frequently used or require sustained activity, such as those involved in eye movement, facial expression, and swallowing. These muscles are often the first to exhibit symptoms because they rely heavily on a high density of functional AChRs to maintain performance. As the disease progresses, larger muscle groups, such as those in the limbs and trunk, may also be affected, leading to generalized weakness. The variability in symptom presentation and severity in MG can be attributed to the degree of receptor destruction and the specific muscles involved.
Understanding antibody-induced receptor destruction is crucial for developing targeted therapies for MG. Treatments such as acetylcholinesterase inhibitors, which increase the availability of acetylcholine at the NMJ, can temporarily improve muscle strength by compensating for the loss of AChRs. Immunosuppressive therapies, including corticosteroids and biologics that target B cells or antibody production, aim to reduce the formation of autoantibodies and slow the destruction of receptors. In severe cases, plasmapheresis or intravenous immunoglobulin (IVIG) may be used to remove or neutralize circulating autoantibodies, providing rapid relief from symptoms. By addressing the root cause of muscle weakness—the destruction of AChRs—these interventions can significantly improve the quality of life for individuals with MG.
Bug Bites and Muscle Spasms: Is There a Link?
You may want to see also
Explore related products






![]()
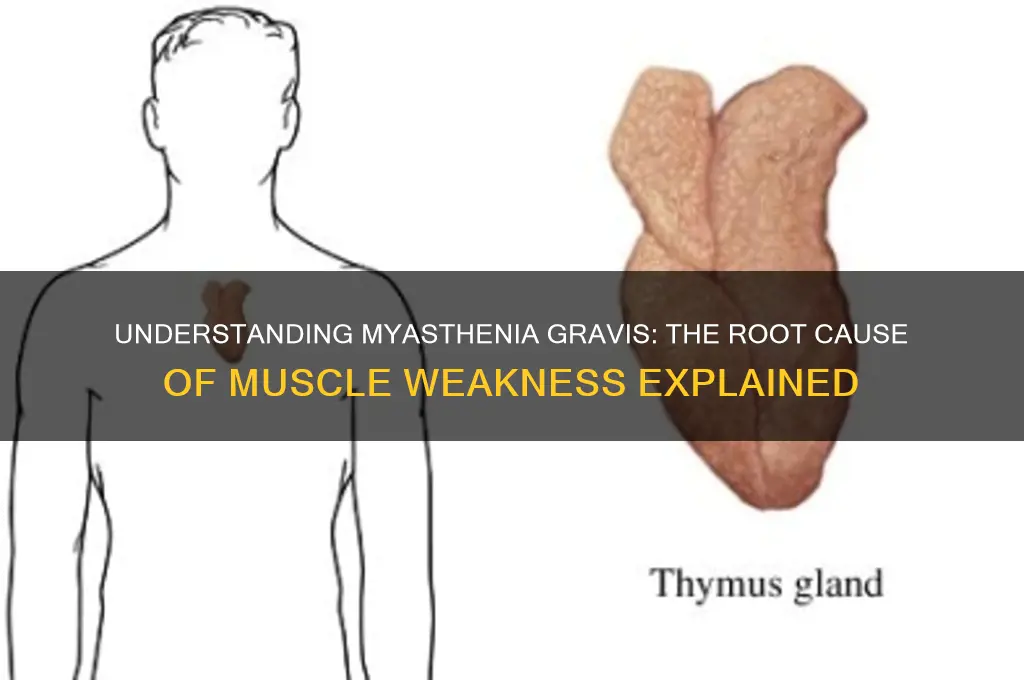
Thymus Gland Abnormalities Role
Myasthenia gravis (MG) is an autoimmune disorder characterized by muscle weakness and fatigue, primarily due to impaired communication between nerves and muscles. Central to the pathogenesis of MG is the abnormal role of the thymus gland, a pivotal organ in the immune system. The thymus, located in the chest, plays a critical role in the development and maturation of T-lymphocytes (T-cells), which are essential for immune function. In individuals with MG, the thymus often exhibits abnormalities, such as hyperplasia (enlargement) or the presence of germinal centers, which are typically associated with immune responses to foreign antigens. These abnormalities are believed to contribute significantly to the autoimmune attack on the neuromuscular junction (NMJ), the site where nerve signals are transmitted to muscles.
One of the key mechanisms linking thymus gland abnormalities to MG involves the production of autoantibodies against acetylcholine receptors (AChR). In a healthy thymus, self-reactive T-cells are eliminated through a process called negative selection, preventing them from attacking the body's own tissues. However, in MG patients with thymic abnormalities, this process is disrupted. The thymus may inappropriately allow the survival and activation of autoreactive T-cells that target AChR. These T-cells then stimulate B-cells to produce antibodies against AChR, leading to their destruction or dysfunction at the NMJ. This disruption in signal transmission between nerves and muscles results in the characteristic muscle weakness of MG.
Thymic abnormalities in MG are not limited to AChR autoantibodies. In some cases, particularly in thymoma-associated MG (a tumor of the thymus), the thymus may produce other autoantibodies, such as those targeting muscle-specific kinase (MuSK) or low-density lipoprotein receptor-related protein 4 (LRP4). These autoantibodies further impair neuromuscular transmission, exacerbating muscle weakness. The presence of thymomas also highlights the direct role of the thymus in MG, as the tumor itself can disrupt normal thymic function and promote autoimmune responses.
The thymus gland's role in MG is further supported by the therapeutic benefits of thymectomy, a surgical procedure to remove the thymus. Thymectomy has been shown to improve symptoms and reduce the need for immunosuppressive medications in many MG patients, particularly those with thymic hyperplasia or thymoma. This improvement is thought to occur because removing the abnormal thymus eliminates the source of autoreactive T-cells and reduces the production of autoantibodies. Additionally, thymectomy may restore normal immune tolerance mechanisms, preventing further damage to the NMJ.
In summary, thymus gland abnormalities play a central role in the pathogenesis of myasthenia gravis by fostering the development of autoreactive T-cells and autoantibodies that target the neuromuscular junction. These abnormalities disrupt nerve-muscle communication, leading to muscle weakness and fatigue. Understanding the thymic contribution to MG has not only advanced our knowledge of the disease but also guided effective treatment strategies, such as thymectomy, to mitigate its impact.
Understanding Left Arm Muscle Pain: Common Causes and Remedies
You may want to see also
Frequently asked questions
Myasthenia gravis is an autoimmune disorder where the immune system mistakenly attacks the neuromuscular junction, the site where nerve signals tell muscles to contract. This disrupts communication between nerves and muscles, leading to muscle weakness and fatigue.
Muscle weakness in myasthenia gravis worsens with activity because repeated muscle use depletes the limited supply of acetylcholine receptors at the neuromuscular junction, which are already compromised by the autoimmune attack. This results in progressively weaker muscle contractions.
Myasthenia gravis typically affects voluntary muscles, especially those controlling eye and eyelid movement, facial expressions, chewing, swallowing, and breathing. Limb muscles can also be involved, leading to generalized weakness.
Muscle weakness in myasthenia gravis is often fluctuating and can improve with rest or treatment. However, without proper management, it may become more severe or persistent. Early diagnosis and treatment are key to preventing long-term muscle damage.