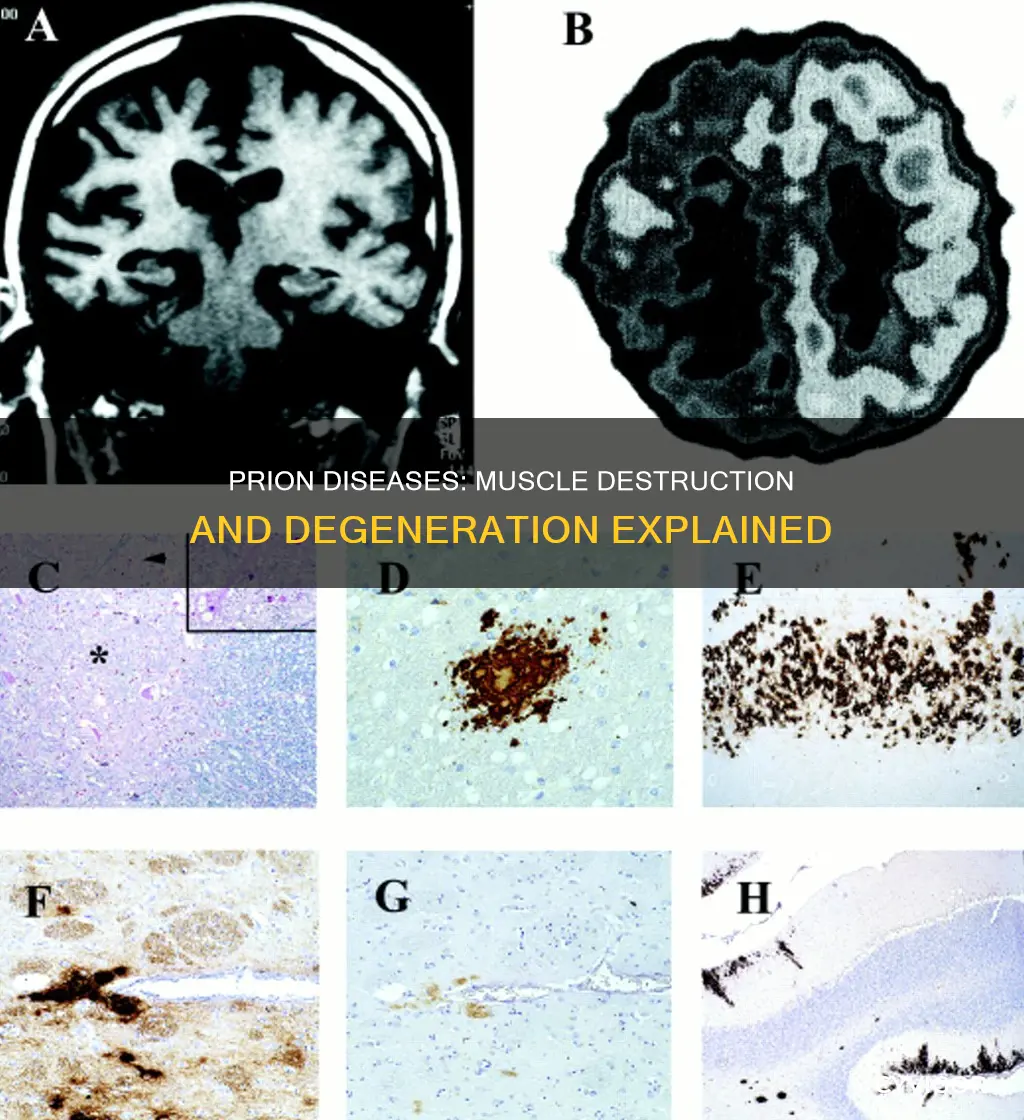
Prion diseases are a group of rare, terminal neurodegenerative diseases that affect both humans and animals. They occur when normal proteins in the brain turn into abnormal proteins known as prions, which cause brain damage and other symptoms like memory loss, movement disorders, and personality changes. While prion diseases primarily affect the brain, recent studies have found evidence of prions in the muscle tissue of food animals, particularly sheep with the prion disease scrapie. This discovery raises concerns about the potential risk of prion transmission to humans through the consumption of meat, even if it is largely free of neural and lymphatic tissue. However, it is important to note that the amounts of prions found in muscle tissue are significantly lower than those in the brains of infected animals, and there is currently no evidence of natural occurrence in bovine muscle tissue.
Do Prions Destroy Muscles?
| Characteristics | Values |
|---|---|
| Prion diseases | A group of rare, terminal neurodegenerative diseases |
| Prions | Abnormal proteins that cause brain damage and dementia |
| Prion sources | Meat from infected animals, organ transplants, contaminated equipment during surgery |
| Prion transmission | Oral ingestion of infected tissues, exposure to contaminated equipment |
| Prion symptoms | Memory loss, thinking and judgment issues, movement disorders, muscle twitching, personality changes, psychological issues |
| Prion diagnosis | Blood tests, lumbar puncture, brain MRI, RT-QuIC test |
| Prion treatment | No cure or treatment, medication to manage symptoms, research on early diagnosis and stopping abnormal protein development |
| Prion prevention | Regulations for handling and feeding cows, excluding certain animal tissues from the human food supply |
| Prion occurrence in muscles | Found in sheep and mouse skeletal muscle, but much lower levels than in the brain |
Explore related products






What You'll Learn
![]()
Prion diseases are rare but fatal
Prion diseases are a group of rare, terminal neurodegenerative diseases that affect both humans and animals. They occur when the normal prion protein (PrPC) in the brain turns into an abnormal misfolded form (PrPSc) known as prions. This abnormal prion then clumps together with other normal prion proteins or binds with them to form more prions. Over time, these abnormal prions damage or destroy nerve cells in the brain, leading to a loss of brain function.
Prion diseases are always fatal and there is currently no cure or treatment. Once symptoms start, the disease progresses quickly, usually within a year. The most common form of prion disease that affects humans is Creutzfeldt-Jakob disease (CJD), which accounts for about 300 cases reported each year in the US. Other prion diseases include variant CJD, kuru, variably protease-sensitive prionopathy, sporadic fatal insomnia, and inherited forms caused by genetic mutations.
The mechanism of prion replication has implications for drug design. Since the incubation period of prion diseases is so long, an effective drug does not need to eliminate all prions but simply needs to slow down the rate of exponential growth. Models predict that the most effective way to achieve this is to find a drug that binds to fibril ends and blocks them from growing any further.
Prion diseases are difficult to diagnose, and confirmation typically requires taking a sample of brain tissue during a biopsy or after death. However, due to the risks associated with brain biopsies, healthcare providers often rely on other tests, such as blood tests, lumbar puncture, and brain MRI, to help diagnose prion diseases. Researchers are also working on developing early diagnostic tools and treatments for prion diseases.
Creatine and Muscle Bloat: What's the Real Deal?
You may want to see also
Explore related products






![]()
Prion diseases are caused by abnormal proteins in the brain
Prion diseases are a group of rare, terminal neurodegenerative diseases. They are caused by abnormal proteins in the brain, which can affect both humans and animals. Prion diseases are always fatal and currently have no cure or treatment. However, certain medications may help slow their progress.
Prion diseases occur when the normal prion protein (PrPc) in the brain turns into an abnormal misfolded form (PrPSc) known as prions. These abnormal prions clump together and bind with normal prion proteins to create more prions. Over time, they damage or destroy nerve cells in the brain, leading to a loss of brain function. Prion diseases can cause a range of symptoms, including memory loss, thinking and judgment issues, movement disorders, personality changes, psychological issues, and more.
The most common form of prion disease in humans is Creutzfeldt-Jakob disease (CJD). It can be inherited genetically or acquired through exposure to infected tissue, such as during a medical procedure. Other types of prion diseases include variant CJD, which is related to "mad cow disease" and can be contracted by consuming infected meat products. Variably protease-sensitive prionopathy (VPSPr) is an extremely rare form of prion disease that affects older individuals with a family history of dementia. Gerstmann-Sträussler-Scheinker disease (GSS) is another rare form that typically occurs at an earlier age. Kuru, seen in New Guinea, is caused by consuming human brain tissue contaminated with infectious prions, but increased awareness has led to a decrease in its prevalence.
Prion diseases are challenging to diagnose due to their rarity and the difficulty of performing brain biopsies. However, healthcare providers employ various tests, such as blood tests, lumbar puncture, and brain MRI, to detect prion diseases before they cause irreversible brain damage. While there is currently no cure, researchers are actively working on developing treatments to stop abnormal protein development and prevent normal proteins from becoming abnormal.
Muscle Extension: Elongating Fibers for Growth and Flexibility
You may want to see also
Explore related products






![]()
Prion diseases can affect both humans and animals
Prion diseases are a group of rare, terminal neurodegenerative diseases that can affect both humans and animals. They are caused by the abnormal folding of prion proteins, which can trigger normal proteins in the brain to also fold abnormally, leading to brain damage and a range of neurological issues.
In humans, the most common form of prion disease is Creutzfeldt-Jakob disease (CJD). This disease can be inherited, in which case it is known as familial CJD, or it can develop suddenly without any known risk factors, which is known as sporadic CJD. Most cases of CJD are sporadic and typically affect people around the age of 60. Acquired CJD can be caused by exposure to infected tissue during a medical procedure, such as a cornea transplant, or by consuming meat contaminated with prions. Variant CJD, for example, is related to "mad cow disease" and can be caused by eating diseased meat. Other rare forms of prion diseases in humans include Gerstmann-Sträussler-Scheinker disease (GSS) and Kuru, which is found in New Guinea and is caused by the consumption of contaminated human brain tissue.
In animals, prion diseases are known as transmissible spongiform encephalopathies (TSE) due to the microscopic changes they cause in the central nervous system and their ability to transmit among susceptible individuals. The first animal prion disease, scrapie, was identified in a merino sheep in Spain in 1732 and has since been found in sheep from Great Britain, goats, and moufflons. Another well-known animal prion disease is bovine spongiform encephalopathy (BSE), which affects cows and was first identified in the mid-1980s. BSE gained widespread attention in the 1990s when millions of cows were infected in the United Kingdom, leading to strong changes in animal production. It has also been linked to variant CJD in humans, raising serious public health concerns. Chronic wasting disease (CWD) is another prion disease that affects deer, elk, moose, and reindeer and has been found in North America, Europe, and Asia. While CWD has not been reported in humans, it is believed to pose a theoretical risk, particularly to those who consume meat from infected animals.
Prion diseases are difficult to diagnose and currently have no cure or effective treatment. They progress rapidly and lead to severe disability and death. However, certain medications can help slow their progress and manage symptoms. Researchers are also working on developing early diagnostic tests and studying ways to prevent abnormal protein development.
Understanding the Role of Quad Muscles as Stabilizers
You may want to see also
Explore related products






![]()
There is no cure for prion diseases
Prion diseases are a group of rare, terminal neurodegenerative diseases that cause brain damage and dementia. They are caused by the normal prion protein (PrPC) in the brain turning into an abnormal misfolded form (PrPSc) known as prions. Prions are small infectious protein particles that induce the "prion protein" to fold abnormally. These abnormally folded prion proteins aggregate and form long, insoluble clumps known as fibrils, which can trigger the death of neurons in the nervous system. This results in different types of neurodegenerative diseases with extensive neuronal loss, including Creutzfeldt-Jakob disease (CJD), the most common form of prion disease affecting humans.
Prion diseases are always fatal and there is currently no cure or treatment to slow down the progression of the disease. However, certain medicines may help alleviate symptoms and slow the progression of the disease. Treatment focuses on keeping patients as safe and comfortable as possible, despite the worsening and debilitating symptoms. Antiseizure drugs or muscle relaxants can be used for myoclonus (muscle twitch), and opioids can be prescribed for pain. The RT-QuIC test can help detect prion diseases before they cause irreversible brain damage, but the disease progresses quickly, making it hard for patients to care for themselves. As a result, patients may need to move to a care facility as the disease worsens.
The mechanism of prion replication has implications for drug design. Since the incubation period of prion diseases is so long, an effective drug does not need to eliminate all prions but simply slow down the rate of exponential growth. Models predict that the most effective way to achieve this is to find a drug that binds to fibril ends and blocks them from growing any further. Some autophagy-promoting compounds have shown promise in animal models by delaying disease onset and death.
The rarity of prion diseases may contribute to the lack of research and treatment options compared to other infectious diseases. Additionally, prions are difficult to destroy as they are resistant to proteases (enzymes that degrade proteins) and are not affected by ultraviolet irradiation, making prevention and decontamination challenging. Furthermore, the precise structure of prions is still unknown, and experts don't know a lot about how they work and are transmitted.
The Truth About Joey Sergo's Muscles: Real or Fake?
You may want to see also
Explore related products






![]()
Prion diseases can be transmitted through infected meat
Prion diseases are a group of rare, terminal neurodegenerative diseases that affect both humans and animals. They occur when the normal prion protein (PrPC) in the brain turns into an abnormal misfolded form (PrPSc) known as prions. Prions are a type of protein that can trigger normal proteins in the brain to fold abnormally, causing brain damage and dementia. While prion diseases cannot be cured, certain medicines may help slow their progress.
The transmission of prion diseases through meat consumption is not limited to humans. The spread of BSE in cattle in the 1980s and 1990s was likely due to feeding practices, with cows being fed meat and bone meal from other infected cows. Another prion disease, Chronic Wasting Disease (CWD), primarily affects wild and captive cervids, including deer, elk, moose, and reindeer. While there have been no reported cases of CWD in humans, experts believe it poses a theoretical risk, particularly to those who consume meat from infected animals.
In addition to meat consumption, prion diseases can be transmitted through other means, such as organ transplants, exposure to contaminated equipment during surgery, or direct contact with infected animals. The rarity and difficulty in diagnosing prion diseases pose significant challenges, and the long incubation periods of prions make detection and decontamination difficult. However, newer regulations governing the handling and feeding of cows, as well as improved management practices, have helped address the immediate threat posed by BSE.
Understanding Paralysis: Muscle Function Loss Explained
You may want to see also
Frequently asked questions
Prions are abnormal proteins that cause brain damage and neurodegenerative diseases. Prion diseases occur when normal proteins in the brain turn into abnormal proteins, which then clump together and cause brain damage and other symptoms.
Prion diseases can cause hypokinetic movement disorders, where an individual moves more slowly than usual or as if their muscles are stiff. Prions have also been found in the muscle tissue of sheep and mice, though the levels of prions in the muscles are much lower than in the brain.
Yes, prion diseases are always fatal and there is currently no cure or treatment. Prion diseases are rare but progress rapidly and lead to death within months to years of the onset of symptoms.
Prion diseases can be contracted by eating meat from infected animals, handling contaminated meat, or through organ transplants or exposure to contaminated equipment during surgery.































